
The biological activity of a protein is determined not only by its static, three-dimensional structure but also by its dynamic characteristics, which form a variety of shapes. This study demonstrates that machine learning can be taught on protein simulation data to build physically realistic conformational ensembles without requiring expensive sampling. The researchers trained a generative adversarial network using coarse-grained simulations of intrinsically disordered peptides, yielding the idpGAN model. This method may overcome computational limits and permit the examination of more extensive and more complicated protein systems.
Examining dynamic characteristics of proteins
The biological activity of proteins is dependent on their dynamic characteristics, which are difficult to examine experimentally. Computational methods, like molecular dynamics (MD) simulations, may be used to construct structural ensembles of proteins; however, the high dimensionality and kinetic barriers provide substantial computing problems. Thus, new methodologies are required to speed the production of dynamic protein ensembles with biological relevance.
Data-driven machine learning algorithms, such as AlphaFold 2, may predict the three-dimensional structure of a protein based on its amino acid sequence, but they do not represent the dynamic character of proteins, particularly intrinsically disordered proteins. Once trained, generative models based on neural networks may rapidly produce statistically-independent samples from the probability distribution of conformations, so bypassing the sampling bottleneck of molecular dynamics. This method can expedite the production of protein conformational and dynamic ensembles.
Generative models and idpGAN
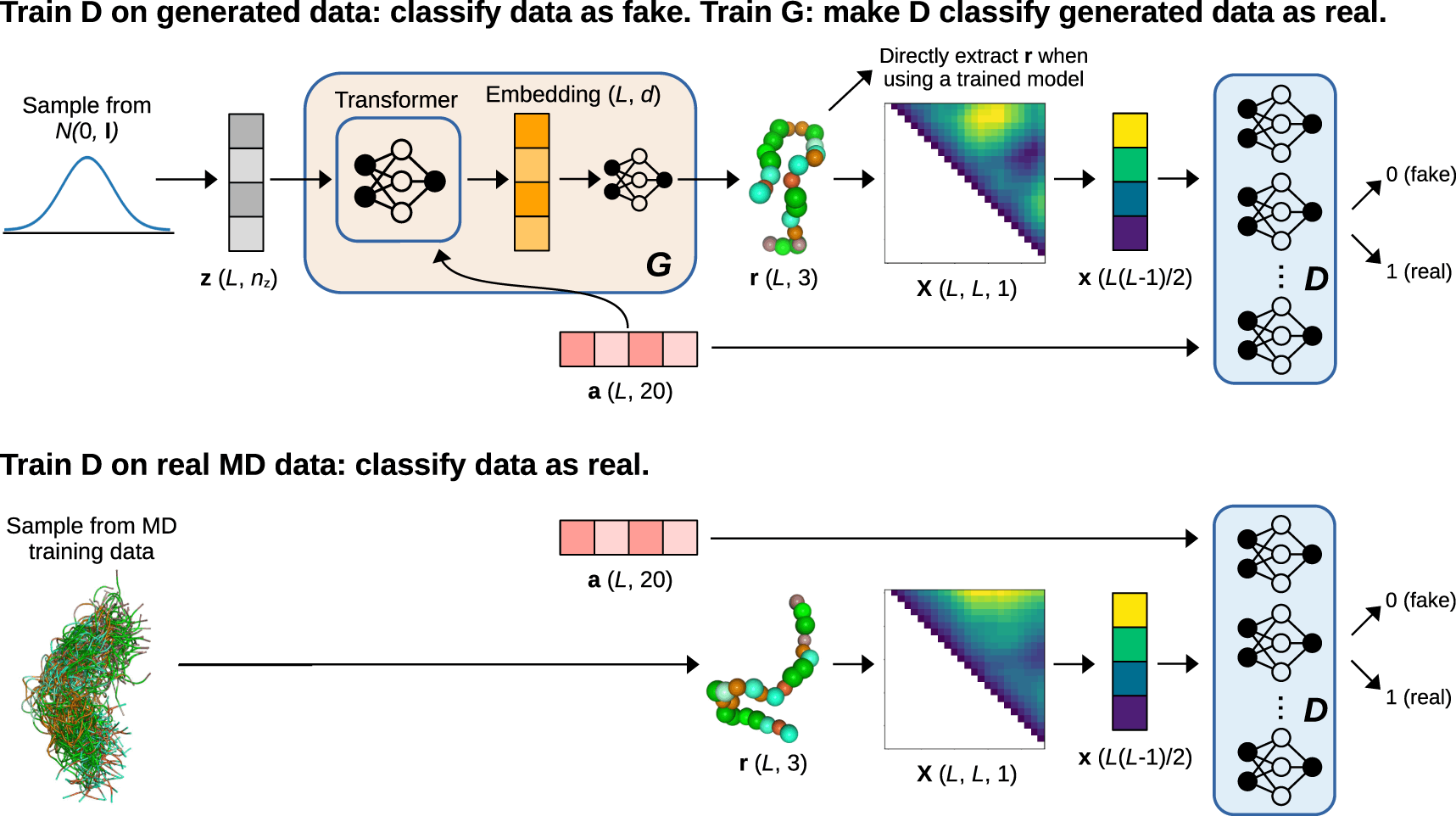
Conditional generative models are used to ensure that generative models can create previously undiscovered molecular conformations for a given system and new systems with alternative chemical compositions. These models are trained with data from several molecules, utilizing the composition of those molecules as conditional information. In this study, ensembles of Intrinsically Disordered Proteins were modeled using a Generative Adversarial Network (GAN)-based conditional generative model called idpGAN (IDPs). GANs are generative models that feature an adversarial game between two neural networks, a generator, and a discriminator, and are capable of producing high-quality samples with rapid sampling capabilities.
idpGAN is a generative model that uses GANs to directly produce C coarse-grained 3D molecular conformations. The G network is built on a transformer design that utilizes a latent sequence and an amino acid sequence as inputs to construct the protein’s 3D coordinates. The D network drives G to create data distributed similarly to the training set. It accepts a protein conformation and an amino acid sequence as inputs and produces a scalar number representing the chance that the combination is real. In the CG MD data, MLPs are employed as discriminators for proteins with varied lengths, but in the ABSINTH data, a single 2D convolutional network discriminator with zero padding is used.
Training the model
The researchers trained idpGAN using residue-based protein data, which was then tested on coarse-grained molecular dynamics data for 31 IDPs not in the training set. The produced conformations were found to be qualitatively realistic and closely matched the ensemble characteristics and energy distributions of the MD-derived ensembles. In addition, the study indicated that idpGAN could capture residue-specific interaction patterns and produce reasonable approximations of MD ensembles, but polyAla chains exhibited distinct discrepancies.
Evaluating the performance of idpGAN
idpGAN’s performance in producing ensembles of intrinsically disordered proteins was evaluated based on various measures (IDPs). The metrics compare the ensembles created by idpGAN to reference ensembles generated by simulations of molecular dynamics (MD) and baseline ensembles generated by a random polymer (polyAla) and snapshots from a different long MD trajectory. In addition to contact maps, the metrics contain pairwise and multidimensional distributions of interatomic distances. The results demonstrate that ensembles formed by idpGAN are typically closer to the reference MD ensembles than the baseline ensembles, notwithstanding some divergence in the most rigorous measure of multidimensional joint distributions. The authors propose that this disparity might be the result of insufficient sampling in the reference MD ensemble rather than poor performance by idpGAN.
To compare the energies of the produced conformations with the reference MD data, the model was evaluated on two test sets, IDP test, and HB_val. As an assessment metric, the median energy differences (MED) between the produced conformations and the reference data were utilized.
The findings demonstrated that idpGAN delivers exceptionally accurate estimates of the MD reference data, with MED values of 5.6 kJ/mol on average for the IDP test set. The model’s performance was shown to be impacted by protein length, with MED values commonly above 50 kJ/mol for proteins with lengths of more than 150 residues, the maximum length employed in the training data. Nevertheless, even with large MED values, the model still represents the distributions of the ensemble’s geometrical characteristics and other aspects. In addition, the findings demonstrated that idpGAN did not overfit on the training data since its performance on the proteins used in its training (HB val set) was only marginally better than on the proteins not used in its training. The constraint of idpGAN with regard to protein length might be circumvented by training with longer crops or by adding a neural-based post-processing refinement phase.
The scientists retrained the idpGAN model with data from all-atom implicit solvent simulations using the ABSINTH potential, which accurately recapitulates empirically known features of several intrinsically disordered proteins (IDPs). ABSINTH simulations are more costly than coarse-grained simulations, thus, training was restricted to peptides with less than 40 residues, and only C atoms were utilized. The retrained model was evaluated on 15 peptides not included in the training set, and for the majority of peptides, idpGAN captured the important characteristics of the reference MCMC ensembles accurately. However, for one peptide, Q2KXY0, the idpGAN ensemble was more disordered than the reference ensemble, likely due to the training set’s emphasis on intrinsically-disordered areas and the GAN model’s inability to simulate more complicated multi-modal ensembles.
idpGAN may model all-atom protein conformational ensembles using explicit solvent simulations, according to the final test. Using prior trajectories, α-synuclein, a big IDP with 140 residues, was tested. idpGAN was adapted to this data by changing the G network and training target. MD ensembles from two separate trajectories validated the model. idpGAN contact maps had the same general structure as MD-based contacts but with considerable variances (MSE_c = 5.78) and some changes in detailed features, indicating overfitting to the training trajectory. The histograms of idpGAN and the training MD data were more comparable than those of the validation MD data, indicating overfitting. The average distance maps were similar (MSE_d = 0.25 nm2), and the distance distributions were correct. KLD_r = 0.30 indicated comparable radius-of-gyration distributions. The results show that an idpGAN-like model trained with adequate trajectory data from varied proteins can generalize to additional sequences.
Improvement in computation speed
One of the primary objectives of idpGAN was to increase computational efficiency compared to conventional approaches such as molecular dynamics simulations. GAN sampling is more efficient since it takes only a forward pass of the G network, which may build ensembles with thousands of conformations in less than 1 second for proteins with lengths of fewer than 150 residues. MD simulations require additional time to obtain the same amount of conformations. While idpGAN can not precisely recover the distributions of long molecular dynamics runs, it can produce near approximations based on KLD_r orders of magnitude quicker than MD. Comparing the sampling efficiency of idpGAN to that of MCMC simulations reveals that idpGAN is substantially quicker.
Conclusion
Without resorting to physics-based iterative sampling, the idpGAN model has the ability to construct realistic conformational ensembles of protein structures directly. It is possible to develop conformations that are energetically advantageous and generate structures for proteins that have not been observed before. Nevertheless, because the produced conformations are independent of one another, kinetic information is lost in the process. If the conformational ensemble is known, it is possible to recover the kinetics using simulation-based re-sampling approaches, which can help overcome this disadvantage.
Article Source: Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Sejal is a consulting scientific writing intern at CBIRT. She is an undergraduate student of the Department of Biotechnology at the Indian Institute of Technology, Kharagpur. She is an avid reader, and her logical and analytical skills are an asset to any research organization.





{kind=link}