

Scientists from the University of Florida, US, and the Research Centre for Natural Science, Hungary, developed a new frame selection algorithm, “ECS-MeDiv,” as an application of the extended continuous similarity indices to describe the conformational diversity in a set of structures.
A fundamental approach to molecular modeling and computational design for the investigation of the dynamics and temporal evolution of molecular systems is known as molecular dynamics (MD).
Since molecular dynamics simulation was the first approach to be effectively ported to the GPU architecture, they have benefited significantly from the exponential rise in computing power that has defined recent decades of computational chemistry research.
Although the size of simulations that can be delivered by new-generation MD software is constantly growing, relatively little work is put into creating post-processing techniques that can keep up with the rapidly increasing volumes of data that are being generated.
In the given study, the scientists provide a novel approach to sampling frames from lengthy MD trajectories that are based on the recently developed extended similarity indices framework.
In contrast to the conventional method of applying a clustering algorithm, which typically scales as a quadratic function of the number of frames, their methodology offers a novel, linearly scaled substitute.
The researchers have seen speedups of up to two orders of magnitude when using it in the case of studies with various system sizes and simulation times when compared to conventional clustering algorithms.
Another benefit for some applications, such as the choice of structural ensembles for ligand docking, is the considerably larger conformational variety of the chosen frames.
Essentiality of MD Simulation and Linear Scaling
The use of molecular dynamics (MD) has emerged as a crucial technique in computational chemistry and related research due to the exponential growth in computer hardware capacity.
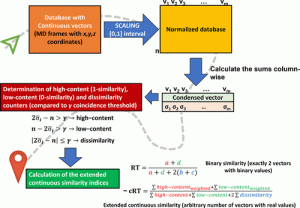
Image Source: https://doi.org/10.1021/acs.jcim.2c00433
Researchers can now model processes down to the microsecond time scale because of the use of graphical processing units (GPUs), which increases the practical length of MD simulations.
While numerous disciplines, including medicinal chemistry, materials science, biophysics, and biochemistry, can benefit from molecular dynamics simulation, this research primarily focuses on the first one.
MD is frequently used to investigate particular molecular events and characteristics, most notably structural alterations, structural stability, chemical interactions, and dynamics of atomic-level phenomena.
By using statistical thermodynamics in conjunction with MD simulations, energies of simulation-related processes can be accounted for as well.
Structures derived from MD trajectories also support other computational techniques. By overcoming the drawbacks of rigid ligand docking, protein structures derived from trajectories make it possible for ligands to fit into a variety of protein shapes.
This strategy, known as ensemble docking, has been demonstrated to improve the efficiency of structure-based virtual screening, a prominent technique for finding early hits in rational drug design.
Furthermore, molecular dynamics simulation can confirm the stability of these protein-ligand complexes.
In addition to structural data, post-processing techniques are needed to retrieve useful information from the simulation output trajectories. Simulation quality analysis, simulation event analysis, and trajectory clustering are frequently used techniques.
To produce representative structures of the given trajectory, the post processing technique is usually used; nonetheless, frame selection and clustering are extremely challenging tasks that might be highly problem-dependent.
The primary concerns are which indices should be used for the selection (the root mean squared distance, or RMSD, is the most popular option) and how the selection should be conducted (most different structures or most common structures).
The majority of commercial MD software packages (such as AMBERtools, Desmond, and NAMD) come with built-in clustering programs; nevertheless, their effectiveness is difficult to assess, and their suitability for various trajectory forms is iffy.
Therefore, the scientists created a diversity picker as an alternative to clustering methods based on the recently released extended continuous similarity indices.
This picker only needs the coordinates of the atoms in the extracted snapshots of the trajectory and is simple to implement.
The diversity pickers, which are frequently used in cheminformatics to sample huge chemical spaces and are typically based on the usage of binary molecular fingerprints, served as an inspiration for the algorithm.
The various versions of the extended similarity indices have shown great promise in the diversity selection and exploration of large and varied data sets, including complex biological ensembles.
The extended indices’ capacity to quantify similarities between any number of items and their ability to do so with linear scaling are the secrets to this achievement.
Circumventing Issues by the Utilization of a Generalization of Extended Similarity Indices
The scientists investigated the categorization of conformations in biological ensembles using extended similarity indices.
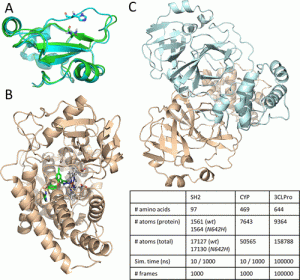
Image Source: https://doi.org/10.1021/acs.jcim.2c00433
To achieve this, they created a brand-new hierarchical agglomerative clustering algorithm that effectively differentiated between conformations corresponding to various folding process phases.
However, because they had to begin with a clustering phase, their method scaled as O (N2). In addition, they only took into account extended similarity indexes constructed from binary vectors.
In other words, a preprocessing step was required to use contact maps to convert the real-valued coordinates into bit-vectors. Three factors make this problematic:
- The preprocessing phase can be time-consuming;
- It is not immediately obvious which residues should be chosen to offer the best contact map representation; and
- There is an inherent information loss when we convert real-valued to binary numbers.
In this study, the scientists define extended continuous similarity indexes to address this latter weakness.
A novel extended similarity-based technique was created to effectively choose a variety of representative structures from extensive MD simulations.
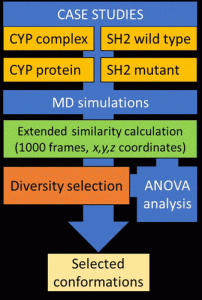
Image Source: https://doi.org/10.1021/acs.jcim.2c00433
In case studies of MD simulations of various lengths and system sizes, they assessed the novel methodology.
The produced trajectories underwent evaluation, and their effectiveness was measured against industry-standard clustering techniques.
To highlight the potential advantages of the extended continuous similarity indices, particularly their good scalability, the devised technique was applied to the post-processing of a 100-second MD simulation of the SARS-CoV-2 major protease.
The post processing is particularly important today since, despite the ability to access simulation durations never before possible because of increased computational capacity and access to powerful supercomputers, post-processing techniques are rarely as effective as the main simulation programs themselves.
Because this novel methodology places more focus on sampling than classification, the various conformations do not require any clustering phase, which makes it particularly appealing since the entire strategy scales as O (N).
Additionally, the researchers avoid all of these problems in this study by employing a modification of the extended similarity indices that work with real-valued values.
This indicates that they can include as many atomic coordinates as possible, that the only preprocessing necessary is a straightforward normalization of the coordinates, and that they are not losing any information throughout the sampling process.
The Endpoint
The scientists looked at four various biologically significant systems with 10 and 1000 ns long MD simulation lengths to investigate the applicability of the suggested methodology.
These systems were the CYP complex, CYP protein, SH2 wild-type, and SH2 mutant. The extended continuous similarity indices were determined for each system and simulation length, and the coordinates of the backbone atoms (C, C, and N) were retrieved for each of the 1000 frames of each simulation.
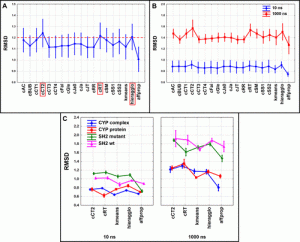
Image Source: https://doi.org/10.1021/acs.jcim.2c00433
For each MD run, sixteen distinct similarity indices were computed and assessed using an ANOVA.
Then, using their diversity picker on the molecular dynamics simulation for each case study, they compared it against industry-standard clustering techniques based on RMSD values.
Finally, using an extraordinarily lengthy MD trajectory of the primary protease (3CLPro) of SARS-CoV-2 with 100,000 frames, the scientists demonstrated how the extended similarity-based diversity selection approach operates.
As a cutting-edge method to describe the conformational diversity in a collection of structures, such as the frames of a molecular dynamics simulation, they have presented the use of extended continuous similarity indices.
Additionally, a brand-new frame selection method dubbed ECS-MeDiv was created, and its performance and applicability were tested against leading-edge alternatives in a variety of frame selection from molecular dynamics simulations.
The extended continuous similarity indices displayed excellent RMSD findings; two of the 16 indices, the cCT2, and cRT, were able to offer a wider range of frame sets than the benchmark techniques.
The ECS-MeDiv, in conjunction with the cCT2 and cRT indices, significantly outperforms the hierarchical agglomerative method when selecting frames from the extraordinarily long 100-second SARS-CoV-2 main protease trajectory as demonstrated by a comparison of the computational requirements for this task with 100,000 frames.
In conclusion, the created cCT2 and cRT-based ECS-MeDiv selection algorithms are appropriate for the wide range of molecular structures retrieved from MD trajectories. The extended continuous similarity indices can be simply modified to characterize the similarity of any data set with continuous values, and their use is completely generic.
Article Source: Molecular Dynamics Simulations and Diversity Selection by Extended Continuous Similarity Indices Anita Rácz, Levente M. Mihalovits, Dávid Bajusz, Károly Héberger, and Ramón Alain Miranda-Quintana Journal of Chemical Information and Modeling 2022 62 (14), 3415-3425 DOI: 10.1021/acs.jcim.2c00433
GitHub: https://github.com/ramirandaq/MultipleComparisons
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}