
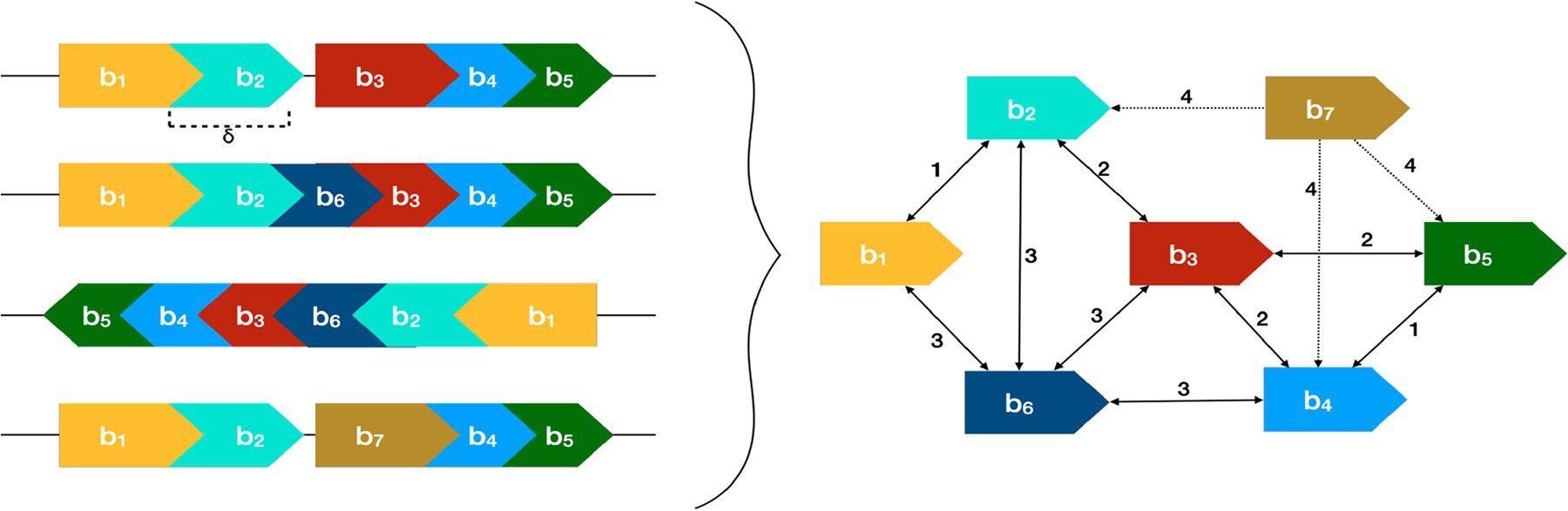
Scientists from the University of Maryland have developed PRAWNS, a scalable and efficient tool for multi-genome analysis. With the unprecedented number of complete and nearly complete genomes being accessible, researchers interested in understanding the genomic basis of phenotypes, such as antibiotic resistance in bacteria, require computational tools capable of performing multiple genome comparisons efficiently. However, currently available computational tools do not scale well in terms of the number of genomes. PRAWNS was developed to fill the gap in the currently available toolbox for understanding genotype-phenotype associations, and this is achieved by defining metablocks, a concise set of genomic features, and pairwise relationships between them.
The need for PRAWNS
Genome sequencing advancements have greatly improved our understanding of genotype-phenotype associations as well as transformed public health and food safety monitoring. Genomic tools for the analysis of small numbers of bacterial genomes are either alignment-based, gene-based, or based on the k-mer approach. The methods are unable to keep up with the pace with which genomes are being sequenced. PRAWNS, on the other hand, have been shown to scale to up to 4,000 genomes while maintaining a concise list of genomic features.
Genomic comparisons enable the characterization of evolutionary events such as horizontal gene transfer, insertions-deletions, and translocations. Genomic variations as such can lead to variations in the phenotype, such as antimicrobial resistance (AMR). Pan-genomic representations aid in the analysis of such genomic variations. Comparative genomics methods involving multiple closely related genomes based on whole-genome alignment include MAUVE, MUGSY, and CACTUS. Gene-focused methods implement gene prediction followed by gene clustering, such as in PANSEQ, PGAP, and ROARY. Other methods like SPLITMEM and TWOPACO construct a colored de Bruijn graph, cdBg, by using exact matching substrings of fixed length k, called k-mers. Whole-genome alignment-based pipeline SIBELIAZ constructs a cdBg using TWOPACO and extracts locally collinear blocks (LCB) using SIBELAZ-LCB.
Bacterial genome-wide association studies (GWAS) for understanding genotype-phenotype relations have been undertaken, and methods have been developed for the same. These methods include SEER and DBGWAS, among others. While these methods are fast enough, they are limited by the loss of genomic context information affecting the results of downstream analysis.
All the above methods have their own share of weaknesses. Alignment-based methods do not scale well with the current number of sequences available (> 100 genomes). Gene-based methods are limited by gene clustering thresholds and gene prediction biases. The cdBg-based methods, although fast, scalable, and not limited to genes, yet produce large numbers of exact matches, which hinders downstream analysis results.
Moreover, mobile elements pose challenges in genome assembly methods and often get placed in separate contigs in draft assemblies. Promoter regions influenced by the mobile elements can lead to changes in gene expression and consequently lead to antimicrobial resistance. Hence, incorporating such genomic variation factors at the pan-genomic level is crucial. Thus, the need for a scalable and efficient algorithm for generating efficient representations of whole genomes that are closely related and provide a succinct list of genomic features shared by a fraction of these genomes. The authors developed PRAWNS to fill this gap.
The two-component algorithmic design of PRAWNS
The input for PRAWNS is a set of whole genomes. For each genome, the unique k-mers are determined and later merged to form shared-exact matching regions called blocks. Next, the neighborhood for each block is obtained. Collated blocks are determined using these neighboring pairs of blocks. Algorithm 1 determines the components of collated blocks by constructing a KNN graph. The collated blocks from each component are merged to form the metablocks. This is achieved by Algorithm 2. In addition, PRAWNS also has a feature to identify paired regions.
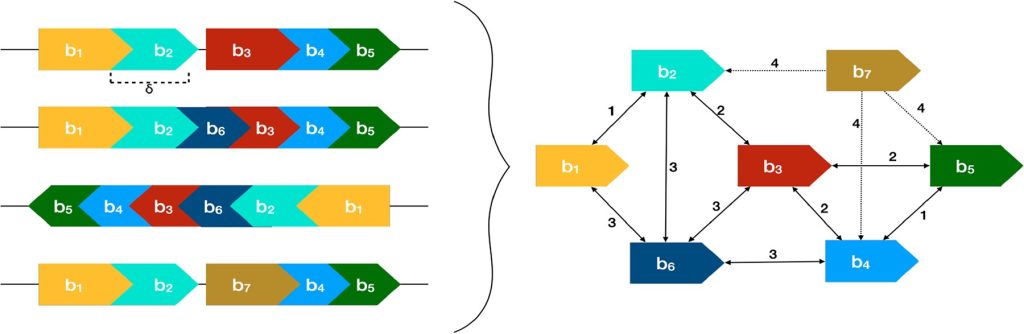
Image source: https://doi.org/10.1093/bioinformatics/btac844
Scalability and efficiency of PRAWNS
The performance of PRAWNS was benchmarked using a whole-genome dataset of 362 Acinetobacter baumannii isolates. Scalability for the method was tested using two datasets, viz, a bacterial and a fungi dataset. PRAWNS was able to decompose conserved regions into metablocks. On the other hand, multiple conserved regions were mapped to a single LCB with SIBELIAZ-LCB, giving rise to redundancy.
PRAWNS was run on a dataset of 4000 bacterial genomes and was executed in 24 hours and yielded a pan-genome with more than 55,000 conserved regions. PRAWNS was also tested on a eukaryotic dataset of fungi that generated a pan-genome comprising more than 85,000 regions in less than 21 hours. PRAWNS was also found to be robust to the choice of k-er length when tested with the bacterial dataset.
AMR in Acinetobacter baumannii: An application
The statistically significant conserved regions, as produced by PRAWNS in this dataset, were found to map to several promoter regions and mobile elements associated with antimicrobial resistance. Understudied antimicrobial resistance-related genes were also mapped to the conserved regions. More than 75,000 paired regions were identified, suggesting the role of the co-presence of genomic factors in antimicrobial resistance.
Conclusion
The need of the hour in comparative genomics was a computational tool for pan-genome analysis. PRAWNS establishes itself as a scalable and efficient candidate to fill this gap in the toolkit for genotype-phenotype association studies. It scales to a large number of genomes and generates a concise set of features with very less computational cost. PRAWNS can be parallelized over multiple threads, and disk-sed storage is used for its computations. However, currently, PRAWNS does not include SNPs and indels. The authors suggest that the metablock structure could be used for identifying such events and plan to include these in future versions. The method promises to be an effective framework for exploring pan-genome genomic variations in bacterial as well as eukaryotic genomes, as in the fungi dataset. Determination of antimicrobial resistance genomic factors would lead to advancement in drug design and better monitoring of food safety and public health.
Article Source: Reference Paper | PRAWNS is freely downloadable from GitHub
Learn More:




Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





{kind=link}