
The emergence of single-cell RNA sequencing (scRNA-seq) has now made it possible to analyze the internal workings of hundreds of cells at once, revolutionizing biology and medicine. However, the limitations of scRNA-seq methods include the potential for inaccurate cell composition analysis and the ineffectiveness of complementary DNA (cDNA) amplification, which is the process by which a double-stranded DNA that “complements” the single-stranded RNA is produced and replicated millions of times by the widely-used template-switching reaction.
A novel and better method for scRNA-seq has just been created by a research team from Japan, led by Assistant Professor Shigeyuki Shichino and Professor Kouji Matsushima of Tokyo University of Science. Compared to the already popular technologies, the new technique, terminator-assisted solid-phase cDNA amplification and sequencing (TAS-Seq) uses less complicated materials and equipment to produce higher-precision scRNA-seq data.
According to Assistant Professor Shichino, the method, TAS-Seq, combines genetic detection sensitivity, the robustness of reaction efficiency, and precision of cellular composition to enable us to capture key cellular information. On June 27, 2022, the study was published in Communications Biology. Associate Professor Satoshi Ueha from Tokyo University of Science, Professor Taka-aki Sato from the University of Tsukuba, and Professor Shinichi Hashimoto from the Wakayama Medical University were also members of the research team.
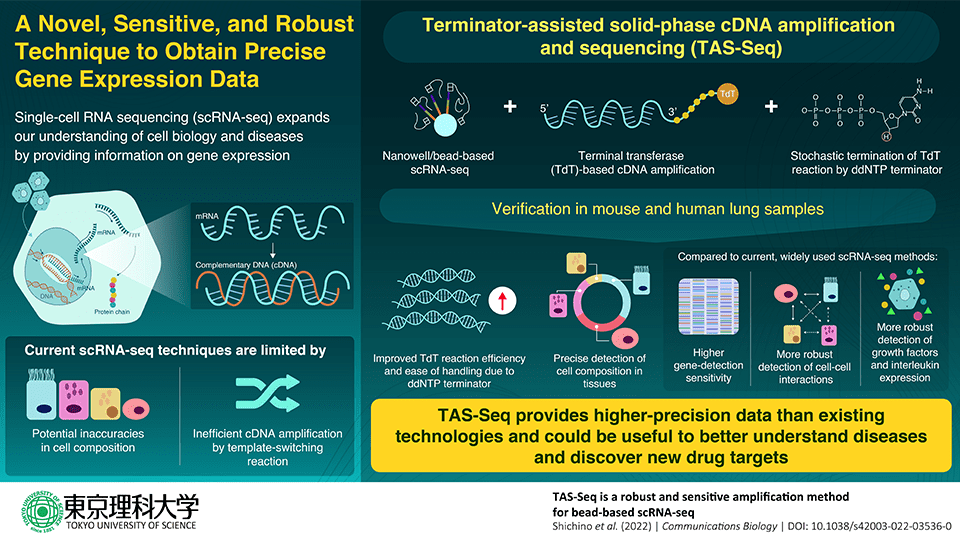
Terminal transferase is a template-independent enzyme used by TAS-Seq to amp up cDNA (TdT). However, TdT is challenging to manage. The research team used dideoxynucleotide phosphate (ddNTP) as a “terminator” for the cDNA amplification step in order to overcome this difficulty. According to Assistant Professor Shichino, ddNTP spike-in, specifically dideoxycytidine phosphate (ddCTP), inhibits the excessive extension of polyN-tail by TdT in a stochastic way and significantly lowers the technical problems of the TdT reaction.
The nanowell/bead-based scRNA-seq platform used by TAS-Seq additionally enables the isolation of single cells from tissue samples, reducing cell sampling bias and enhancing the precision of cell composition data. Using samples of murine and human lung tissue, the study team next confirmed the effectiveness of TAS-Seq and evaluated it against two popular, contemporary scRNA-seq methodologies, 10X Chromium V2 and Smart-seq2. Compared to the most popular scRNA-seq technologies, the researchers discovered that TAS-Seq could detect more genes overall and find more highly variable genes.
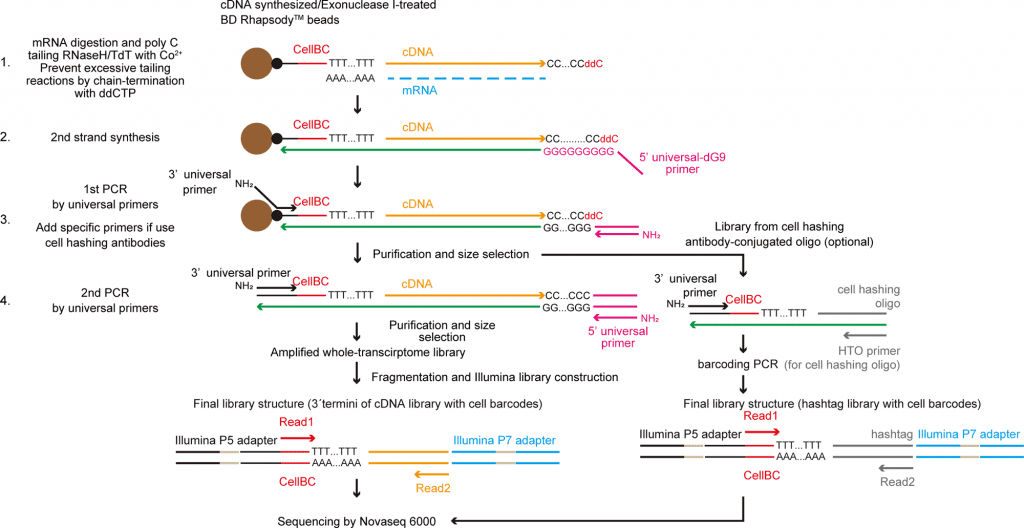
Image Source: TAS-Seq is a robust and sensitive amplification method for bead-based scRNA-seq
According to Assistant Professor Shichino the researchers discovered that TAS-Seq might perform better in terms of gene detection sensitivity and gene drop-out rates than 10X Chromium V2 and Smart-seq2, suggesting that TAS-Seq may be one of the most sensitive high-throughput scRNA technologies. Genes may be detected more evenly over a wide range of expression levels, and growth factor and interleukin gene detection is also improved.
The new technique also has the benefit of TAS-Seq being less prone to batch effects. The fact that TAS-Seq data and flow-cytometric data from the tissue samples had a strong correlation further suggests that TAS-Seq can produce extremely precise cell composition data.
Talking about future research, Assistant Prof. Shichino says the research team has already created TAS-Seq2, an enhanced and highly optimised version of TAS-Seq. In mouse spleen cells, TAS-Seq2 provides 1.5–2 times more sensitive gene detection. In order to offer scRNA-seq services using TAS-Seq and TAS-Seq2, the research team has also founded ImmunoGenetics, a venture firm from the Tokyo University of Science.
For scientists working in medicine and biology, scRNA-seq is a crucial tool. TAS-Seq and TAS-Seq2 will progress the science of “spatial transcriptomics,” which also depends on solid-phase cDNA synthesis by identifying novel therapeutic targets for illnesses. Additionally, it will hasten the advancement of single-cell omics technologies, advancing our comprehension of the biological underpinnings of illness onset and progression.
Story Source: Shichino, S., Ueha, S., Hashimoto, S. et al. TAS-Seq is a robust and sensitive amplification method for bead-based scRNA-seq. Commun Biol 5, 602 (2022). https://doi.org/10.1038/s42003-022-03536-0 https://www.tus.ac.jp/en/mediarelations/archive/20220627_5628.html
Learn More About Bioinformatics:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











{kind=link}