
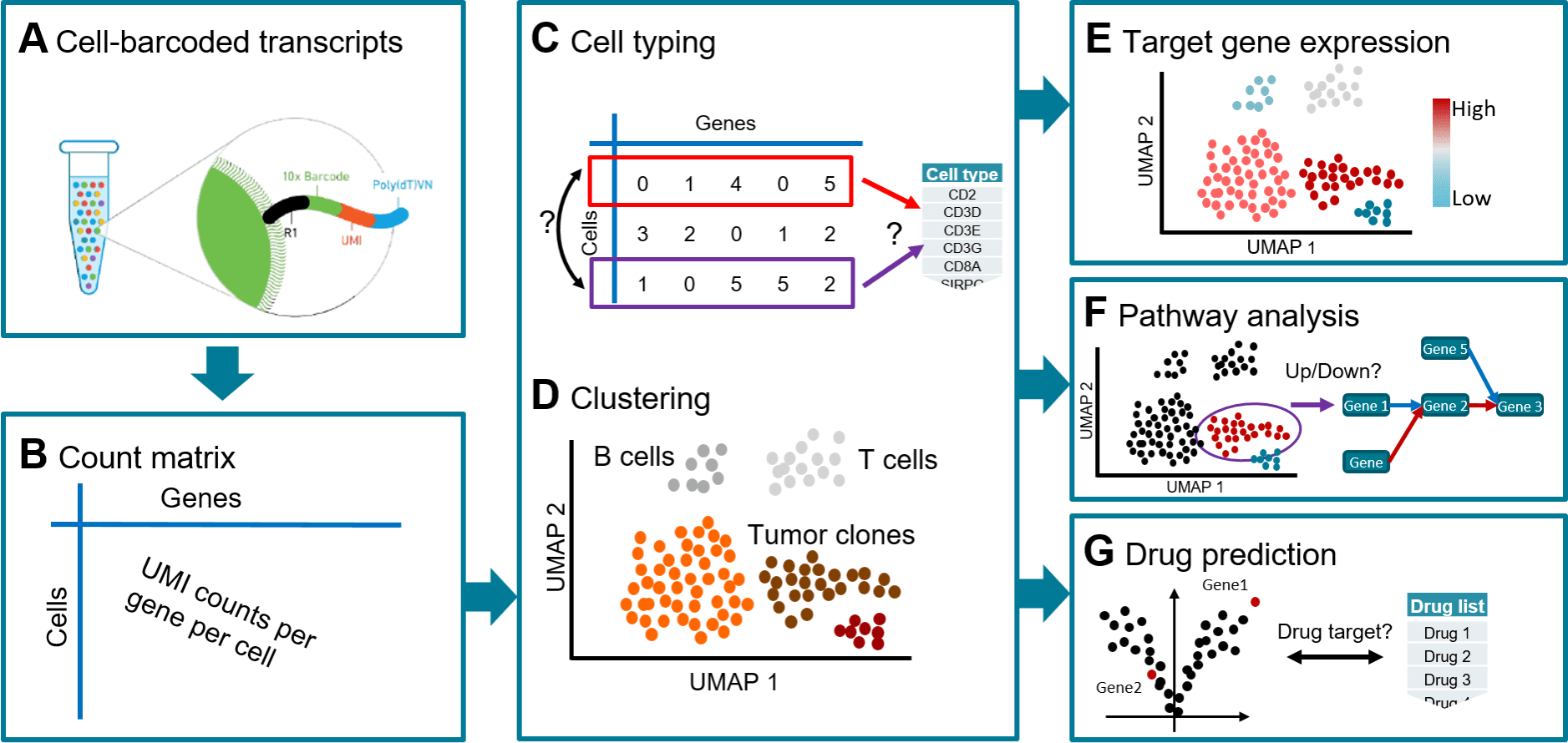
Scientists from the Swiss Institute of Bioinformatics devise a workflow known as Single Cell Analysis mRNA Pipeline, ‘scAmpi,’ for providing complete functionality to scRNA-seq data analysis.
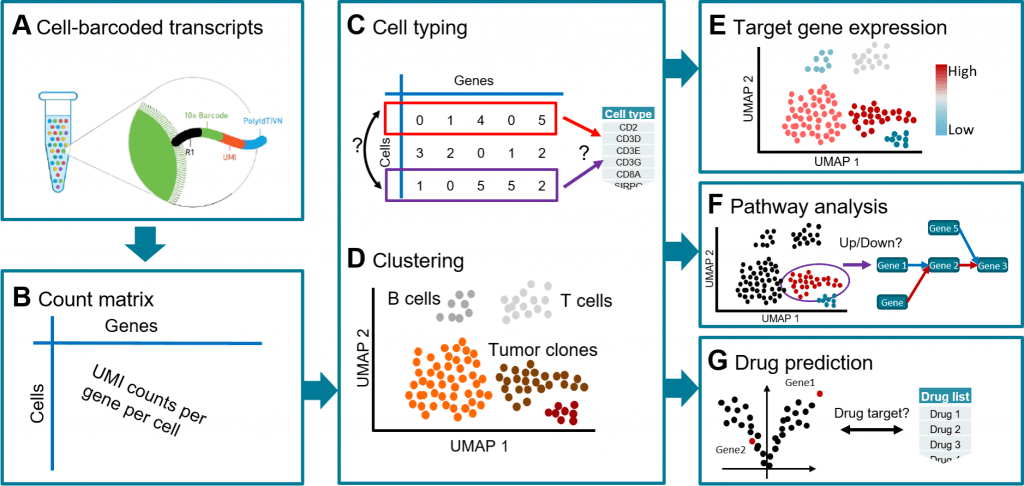
Image Source: https://doi.org/10.1371/journal.pcbi.1010097.g003
In order to understand tissue composition at the single-cell level, to shed light on disease processes, tumor heterogeneity, and the condition of the immune milieu, single-cell RNA sequencing (scRNA-seq) has become an effective tool.
Although there are several computational methods for analyzing scRNA-seq data, their use in a clinical environment necessitates structured and consistent procedures that are designed to extract, condense, and show clinically pertinent information.
In order to achieve this, scientists created the workflow known as scAmpi (Single Cell Analysis mRNA Pipeline), which streamlines the scRNA-seq analysis process from raw read processing to informing on sample composition, clinically relevant gene and pathway alterations, and in silico identification of personalized candidate drug therapies.
In a molecular tumor board as a part of a clinical investigation, they show the value of this procedure for clinical decision-making.
Ineffectiveness of Existing Software in Routine Clinical Use
Single-cell RNA sequencing (scRNA-seq) is a high-throughput technique that has become popular in recent years for determining gene expression at the single-cell level.
It offers previously unattainable insights into topics like cell differentiation, the immune compartment, and tumor heterogeneity, among others.
A growing number of researchers employ scRNA-seq to examine clinical samples such as tumor tissues after it was first used to describe PBMCs or developing stem cells.
For general scRNA-seq analysis, numerous software suites are available, including the widely-used tools SEURAT and ScanPy or the web-based software suites CreSCENT and ASAP. They do, however, have a few drawbacks:
- First, non-bioinformaticians may find it challenging to use the tool because doing so requires at least rudimentary R or Python programming skills.
- Second, no other software enables the discovery of therapeutic candidates in silico using single-cell data.
- Last but not least, current software suites are not suitable for routine clinical use because they are not built to manage large-scale data analysis in a fully reproducible, transparent, and auditable fashion, including error tracking and process documentation.
As a result, scientists created scAmpi, a complete turnkey pipeline for scRNA-seq research that includes processing raw reads and providing information on sample composition, gene expression, and prospective medication candidates.
With the help of the Snakemake workflow management system, scAmpi is simple to use, has a lot of flexibility in the methods that may be used, and can be used in a highly standardized way reproducible.
This has enabled the ongoing Tumor Profiler clinical investigation to successfully use scAmpi for processing scRNA-seq data.
Interpretation of Results from scAmpi
The researchers present the readout and analyses scAmpi can do on scRNA-seq data obtained from a biopsy of a melanoma patient who participated in the Tumor Profiler clinical study.
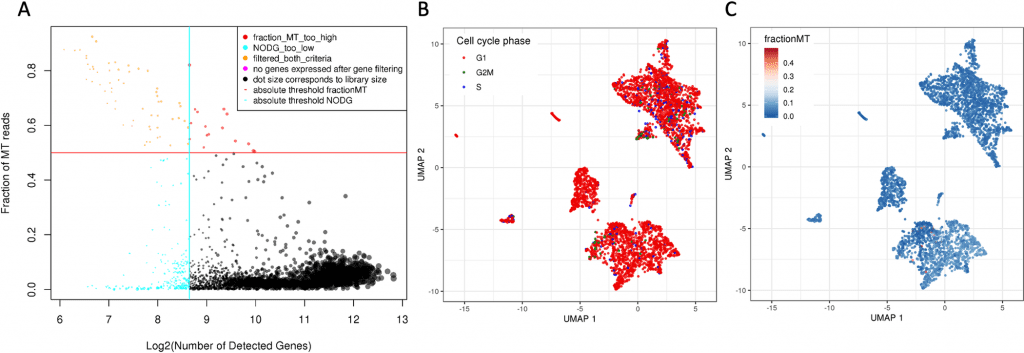
Image Source: https://doi.org/10.1371/journal.pcbi.1010097.g003
With just two instructions, the entire analysis—from the raw fastq files through the identification of medication candidates—can be started.
Cellranger recognizes 4193 cells in the first mapping step. 10% (437) of the cells are removed by subsequent scAmpi screening due to poor quality. The cell-cycle phase appears not to affect the embedding of the cells after normalization.
According to the study of cell types, 34% of the sample’s cells are melanocytic melanoma cells. The immunological microenvironment of tumors is quite varied and contains many different types of cells, including macrophages, B cells, NK cells, endothelial cells, memory effector T cells, and a sizable population of T cells.
This conclusion is consistent with findings from CyTOF tests that were also conducted in a case study. Gene expression visualization, as well as a population-based and ranked overview of the average gene expression and the number of non-zero cells for each gene, assist further research into the immunological milieu.
The melanoma population divides into four clusters, which suggests tumor heterogeneity, according to unsupervised clustering.
To further explore this heterogeneity, scAmpi provides a variety of readouts, including individual gene expression analysis, gene set enrichment analysis, and differential gene expression when comparing tumor clusters.
The use of BRAF/MEK inhibitor therapy is not possible in three of the four tumor clusters due to down-regulation of the MAPK pathway (gene set derived from the Hallmark MSigDB).
The entire tumor population was indicated as possibly responsive to palbociclib treatment by scAmpi’s in-silico drug candidate identification, based on the over-expression of CCND1 and further reinforced by the expression of CDK4.
This result has been seen with various technologies, such as drug response testing.
When combined, scAmpi offers information on the overall sample makeup, gene expression, and pathway, as well as the ability to analyze downstream data to aid in clinical decision-making.
scAmpi: Future Scope
scAmpi provides complete functionality for scRNA-seq data analysis. The application to numerous tissues and disease types is made possible by the flexibility and simplicity of the method, which are key features.
However, it offers a consistent and repeatable procedure that may be applied in clinical settings and has already been used in a clinical trial.
Image Source: https://doi.org/10.1371/journal.pcbi.1010097.g003
Additionally, scAmpi makes it easier to identify drug candidates in silico at the single-cell level, directly accounting for disease heterogeneity in the formulation of the best medication therapy.
The pipeline and its open source code will also continue to be improved by the single-cell community due to the modular Snakemake foundation.
The Endpoint
The scRNA-seq, or single-cell RNA sequencing, quantifies the levels of gene expression in each cell.
As a result, it is well adapted to provide information on cell type composition and activity in various tissues and illnesses.
However, it might be difficult to properly handle and interpret the substantial volumes of data produced by scRNA-seq.
In order to achieve this, the scientists created the scAmpi (Single Cell Analysis mRNA pipeline), an analysis workflow that starts with the raw sequencing data and performs preprocessing quality control and subsequent analysis stages in accordance with cutting-edge guidelines for scRNA-seq processing.
The approach eliminates cells with poor quality, labels each cell with its cell type, and displays the expression of certain genes of interest and functional pathways on the individual cells.
Additionally, scAmpi can connect the observed gene expression in disease-related analysis to possible medication candidates that might be effective in treating the disease.
Article Source: Bertolini A, Prummer M, Tuncel MA, Menzel U, Rosano-González ML, Kuipers J, et al. (2022) scAmpi—A versatile pipeline for single-cell RNA-seq analysis from basics to clinics. PLoS Comput Biol 18(6): e1010097. doi:10.1371/journal.pcbi.1010097
GitHub: scAmpi’s Source Code
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}