

Scientists use PandaOmics, an AI-driven target identification tool, to examine the expression profiles of direct iPSC-derived motor neurons from Answer Amyotrophic Lateral Sclerosis (ALS) and central nervous system samples from public datasets.
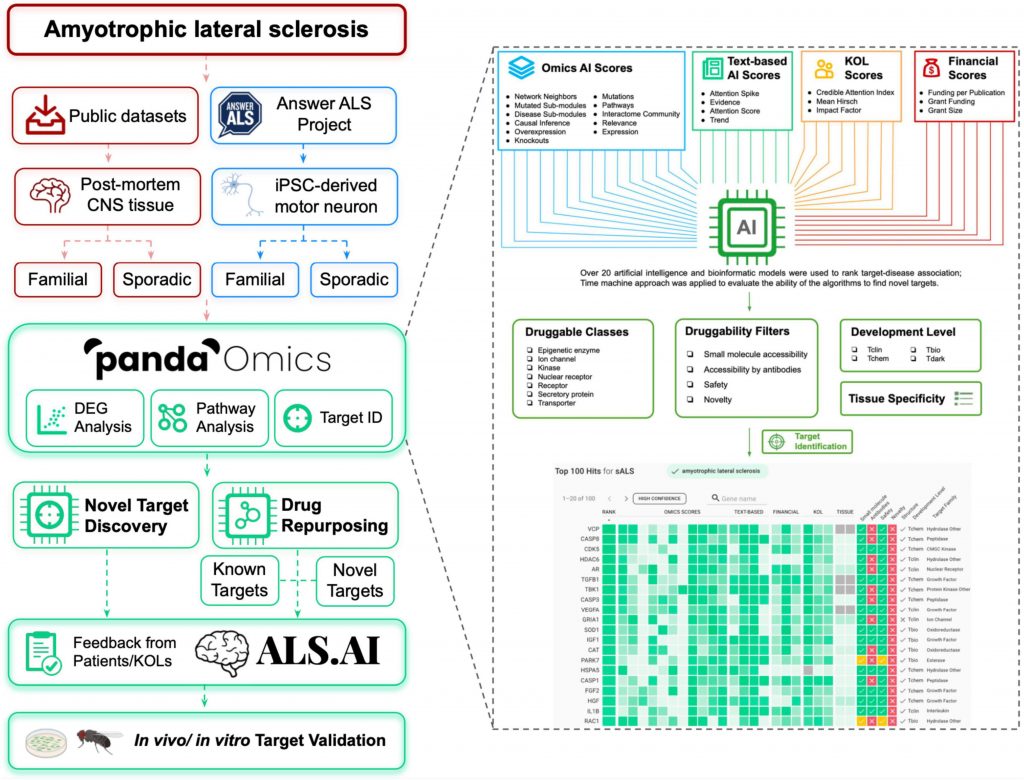
Image Source: https://doi.org/10.3389/fnagi.2022.914017
The discovery of novel therapy regimens is urgently needed for the severe neurological disease amyotrophic lateral sclerosis, which has unclear pathophysiology. Here, the researchers used PandaOmics, an AI-driven platform for target discovery, to assess the expression patterns of central nervous system (CNS) samples from public datasets (237 cases; 91 controls), as well as direct iPSC-derived motor neurons (diMNs) from Answer ALS (135 cases; 31 controls).
On ALS.AI (http://als.ai/), 17 high-confidence therapeutic targets and 11 unique therapeutic targets have been found.
The scientists confirmed eight previously unreported genes (KCNB2, KCNS3, ADRA2B, NR3C1, P2RY14, PPP3CB, PTPRC, and RARA) whose knockdown significantly reverses eye neurodegeneration among the suggested targets evaluated in the c9ALS Drosophila model.
Different stages of illness progression are characterized by dysregulated pathways found in the CNS and diMN data.
Overall, this study adds to the researchers’ understanding of ALS pathogenesis, shows how AI accelerates target discovery, and creates new avenues for therapeutic approaches.
Amyotrophic Lateral Sclerosis and Viability Reduction of Affected Neurons
An uncommon neuromuscular condition called amyotrophic lateral sclerosis is brought on by the progressive degradation of motor neurons (MNs) in the brain and spinal cord.
The Ice Bucket Challenge, which attracted famous people, public figures, and athletes from all around the world, successfully raised awareness of this life-threatening disease in 2014.
According to Roman (1996), it is the most prevalent disease in Minnesota, with an incidence of 0.6 to 3.8 per 100,000 person-years. In the United States, around 16,500 cases of ALS were identified in 2015.
The disease typically begins in middle adulthood, and the average post-diagnosis survival period is around three to five years.
Although the location of the neurons that are damaged varies, the typical signs and symptoms of ALS are gradually progressive muscular weakening and paralysis.
Age-related diseases like ALS are projected to become more common as the population ages. According to studies, aging causes DNA damage, oxidative stress, protein damage, and an accumulation of damaged proteins, all of which lower the survival of the afflicted neurons.
The proposition of Several Pathophysiological Mechanisms
Based on its underlying causes—familial or sporadic—ALS can be divided into different groups.
Ten percent of cases of ALS are caused by familial ALS (fALS), which is caused by mutations in particular genetic loci that are passed down via the autosomal dominant gene.
There are over 20 genetic risk factors for fALS. The thorough characterization of SOD1, TARDBP, C9orf72, and FUS is noteworthy.
Four major ALS-associated genes account for 47.7% of familial ALS (fALS) cases and 5.2 perper cent sporadic ALS (sALS), according to a pooled summary of mutation frequency from 111 studies. However, a significant portion of the genetic basis for ALS remains unknown.
Numerous pathophysiological explanations for ALS have been proposed, taking into account the disease’s diverse genetic involvement.
These explanations include abnormal proteostasis, altered RNA metabolism, nucleocytoplasmic transport defects, mitochondrial dysfunction, DNA repair deficiencies, axonal transport defects, vesicle transport dysregulation, excitotoxicity, oligodendrocyte dysfunction, and neuroinflammation.
Demonstration from the Phase 3 Studies
Due to the researchers’ incomplete understanding of the disease’s processes, ALS is an incurable condition at this time.
Riluzole, Tiglutik, Edaravone, and Nuedexta are the four medications for the treatment of ALS that have received FDA approval in the United States.
The first FDA-approved neuroprotective treatment for ALS and the only medication that increases the longevity of ALS patients is riluzole, an inhibitor of the sodium channel subunit.
Riluzole comes with a wide range of neural effects in addition to blocking glutamatergic transmission, including inhibition of fast and persistent sodium currents, inhibition of neurotransmitter release, reduction of voltage-gated calcium and potassium currents, and potentiation of calcium-dependent potassium current.
These are potential explanations for the medication’s minimal ability to increase patient survival.
Riluzole’s oral suspension formulation, Tiglutik, was created for ALS patients who have trouble swallowing.
Edaravone is a free radical scavenger that prevents MN mortality and inflammation brought on by reactive oxygen species. In Japan, it was authorized for sale and production in 2015, and the FDA approved it as an ALS treatment in 2017.
In clinical studies, edaravone reduced peroxynitrite and its relationship with neurotoxicity in plasma and cerebral fluid of ALS patients, respectively.
Although edaravone-treated ALS patients have shown a significant improvement in their ALS Functional Rating Scale-Revised (ALSFRS-R) score, its long-term efficacy is still debatable and needs more research to be confirmed.
Dextromethorphan and quinidine are ingredients in the oral medicine Nuedexta. Its effectiveness in lowering the frequency and severity of pseudobulbar impact in ALS patients has been shown in phase 3 studies.
Identification of Therapeutic Drugs for ALS
Numerous genome-wide association studies have searched for frequent genetic variations in ALS as a result of developments in genomic profiling techniques, and this research has discovered new candidates as either genetic risk factors or biomarkers (e.g., ACSL5, KIF5A, ATXN2, and MOBP).
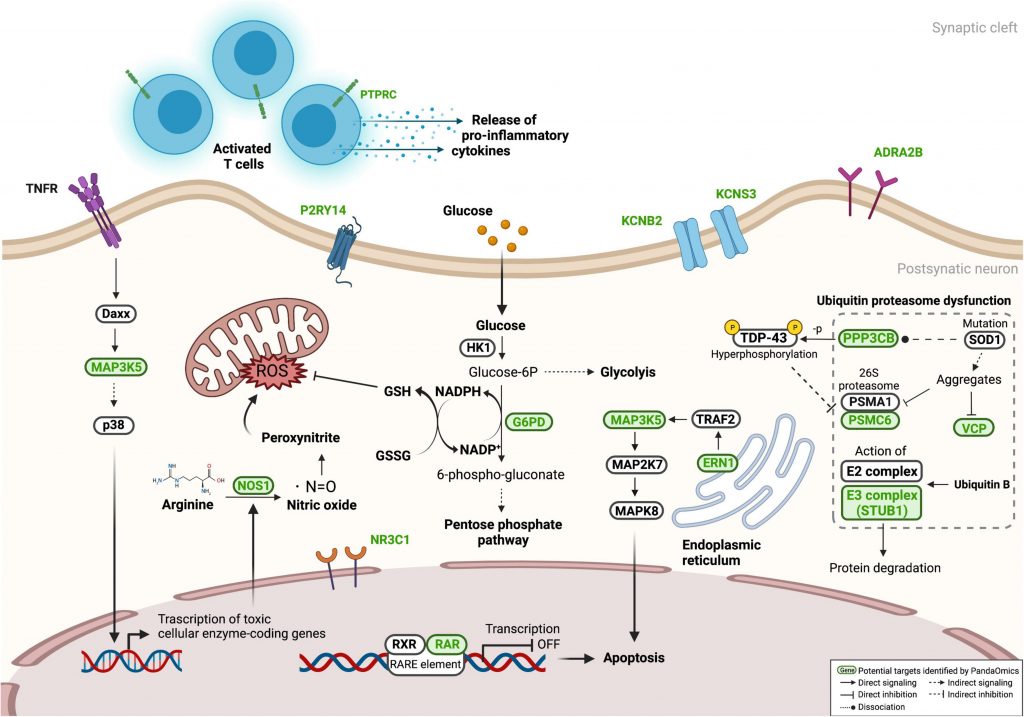
Image Source: https://doi.org/10.3389/fnagi.2022.914017
Finding the differentially expressed genes that contribute to the disease-causing pathways may also be aided by genomic profiling of central nervous system (CNS) tissues and blood from ALS patients.
Additionally, the use of ALS-linked gene variations in both cellular and animal models makes it possible to identify probable interaction partners for those ALS-linked genes, offering additional lines of evidence for identifying disease pathology.
To find prospective treatment targets, the scientists used PandaOmics, an AI-powered target discovery tool, to examine the dysregulated expression of genes and changed pathways across diverse ALS-related datasets.
To conduct target identification, they utilized post-mortem CNS tissues and directly differentiated motor neurons (diMN) obtained from ALS patients.
PandaOmics rates targets based on their target-disease relationships as well as information on druggability, developmental state, and tissue specificity using more than 20 AI and bioinformatics models.
A total of 28 candidates—17 high-confidence and 11 novel—were chosen as prospective ALS therapy targets by adjusting various filter parameters. The ALS.AI platform will receive the proposed targets.
The effectiveness of this strategy was assessed by validating suggested candidates in a Drosophila model that mimicked C9orf72-mediated ALS (c9ALS), the most prevalent form of fALS.
The study’s purpose is to show how to use PandaOmics, an AI-driven target discovery platform, to find therapeutic targets for ALS.
The Endpoint
In conclusion, the given study reveals how to use their most recent AI models based on in-depth omics data analysis to find and prioritize high-confidence and innovative targets for ALS.
Numerous well-known processes, including the immune system, RNA metabolism, excitotoxicity, and programmed cell death, were discovered to be dysregulated in ALS pathogenesis. From CNS and diMN samples, 17 high-confidence and 11 unique therapeutic targets were found.
Results from diMN comparisons are more likely to be linked to the early-stage signatures of ALS, whereas CNS data primarily represents late-stage ALS signatures.
A thorough insight into the course of the ALS disease could be obtained by combining the use of diMN and post-mortem CNS tissues. The use of the Drosophila model demonstrated how to quickly test for AI-identified targets.
The scientists were able to confirm eight previously unreported genes (KCNB2, KCNS3, ADRA2B, NR3C1, P2RY14, PPP3CB, PTPRC, and RARA) whose knockdown significantly prevented eye neurodegeneration among the 26 potential targets evaluated in the c9ALS Drosophila model.
Future research employing diMNs or mammalian models is required to elucidate their pathogenic function and potential as therapeutic targets for c9- and other kinds of ALS.
Targets identified in this study using PandaOmics will be made public on ALS.AI to speed up the discovery of novel targets and therapeutic research for ALS. The report provides fresh perspectives on how AI shortens the target identification process from years to months.
Article Source: Frank W. Pun, Bonnie Hei Man Liu, Xi Long, Hoi Wing Leung, Geoffrey Ho Duen Leung, Quinlan T… Identification of Therapeutic Targets for Amyotrophic Lateral Sclerosis Using PandaOmics – An AI-Enabled Biological Target Discovery Platform Frontiers in Aging Neuroscience, June 2022. DOI
Learn More About Bioinformatics:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}