
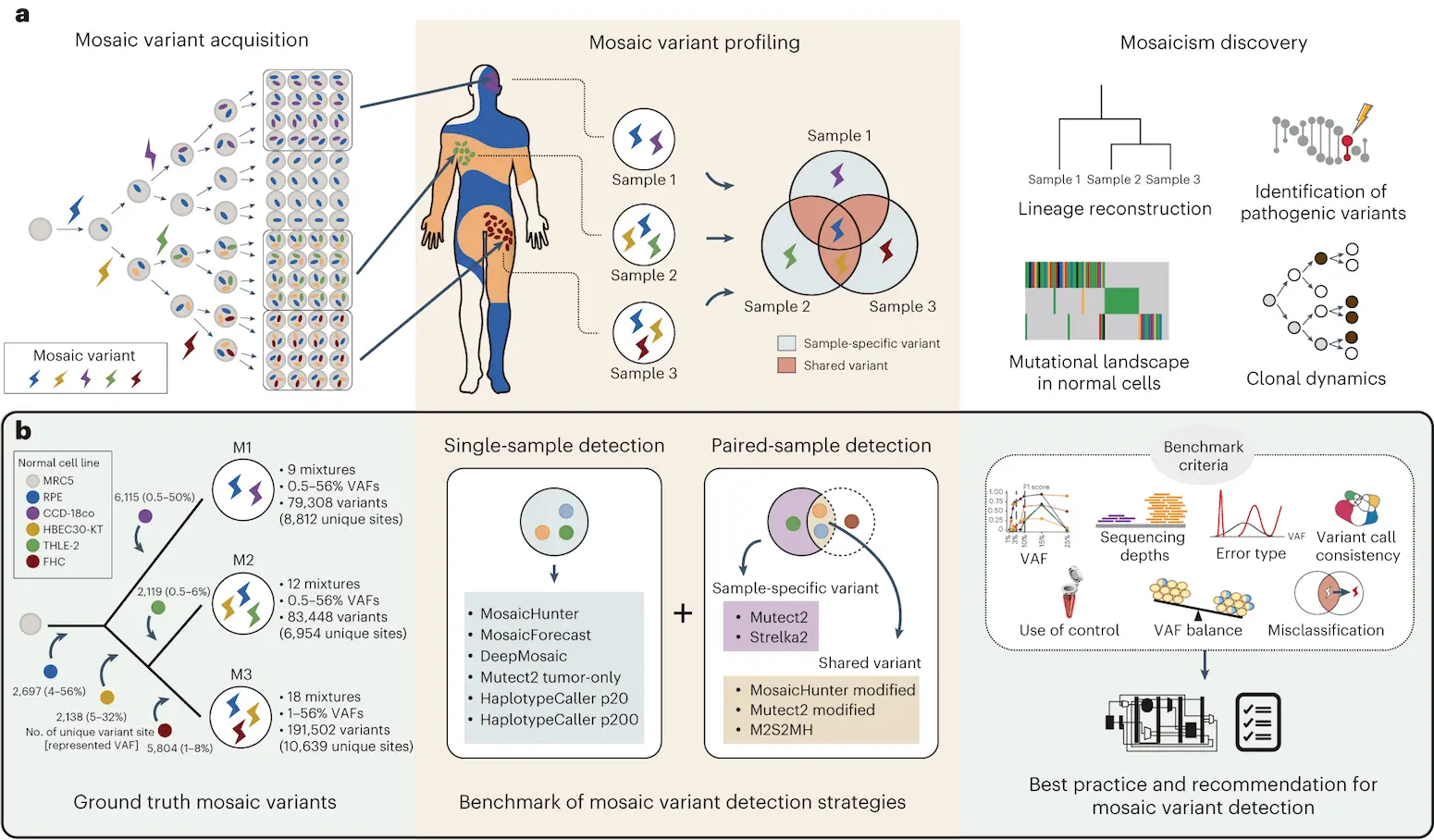
Scientists from Yonsei University College of Medicine, Republic of Korea, have developed a new benchmark for evaluating mosaic variant calling methods, which are used to detect genetic mutations in individuals. The benchmark will help researchers select the most suitable calling algorithms for their studies and guide developers in improving the accuracy of these methods.
The researchers developed 11 different approaches to detect mosaic variants within a genome based on a reference standard that has been designed on the level of a whole exome, the part of the genome consisting entirely of exons. Mosaic variants bring forward a condition where more than one genetic line is present due to genetic mutations. In simple words, two cells within the same organism will have different genetic codes. Detection of heterozygous, homozygous, and mosaic mutations within the genome has been made possible using newly developed sequencing technology for analysis. There have been many technical difficulties in evaluating mosaic variant callings in a conceptual manner as of now, therefore, this field of research still needs to be developed. The best method of detection identified by the researchers found conditional advantages and disadvantages present in existing methods as well. They also found that the evaluation of mosaic variants on a feature level and their usage in the form of combinations act as important factors that contribute to improving their detection.
An introduction to mosaic variants
Genetic mosaicism is a situation caused when the zygote transitions into an ‘adult,’ that is, a defined zygote. A zygote is a diploid, having two complete sets of chromosomes resulting from a fertilized ovum. Postzygotic mutations are a continuous occurrence, and examination of these mutations can help us understand the processes of mutation that contribute to several conditions and developments in aging, cancer, and neurological disorders. Considering the potential, further research in this domain is warranted.
Detecting mosaic variants is a complex process that is initialized through conceptualization. The scope of mosaic mutations is not well defined. Some somatic mutations can also be considered to exhibit mosaicism, as they can show several genetic differences within the same individual. One of the major challenges faced in this domain is that mosaic mutations may occur in more than one tissue; this makes applying controls a lot more complicated.
Parameters used to detect mosaic variants
Variant allele frequencies (VAF) is a parameter that tells us about the frequency of variable alleles within the genome. These frequencies can either be very low or extremely high; the latter is the case in many heterozygous variants. Heterozygous variants imply that both the alleles within a gene differ from each other, whereas alleles within homozygous variants have the same expression. The proportion of mosaic variant occurrences can be imbalanced in terms of their timing and location, as they can be variable.
A combination of different approaches have been taken previously to detect mosaic variants, such as targeting unlikely VAFs that have typically given zygosity within a single sample and searching for shared variants within samples that are shared; both can be enhanced by incorporating machine-learning algorithms as a tool to aid the process. However, a more detailed and well-designed algorithm will be required to cover the full extent of mosaic variants present within the genome.
Existing technologies used for detecting mutation
Some of the approaches that have been used by the researchers in this study are insertion-deletion (INDEL) and single nucleotide variant (SNVs), with both of them based on a reference standard that can be comprehended. Multiple parameters that users may require for conducting scientific analysis have been taken into consideration, such as VAFs, variant sharing and their types, depth of sequences, matched controls, and VAF balances. The strengths and weaknesses of this algorithm, as well as its accuracy, have been evaluated.
For the detection of somatic and germline mutations, many different technologies have been developed over the course of the past few years. This study shows that there are lower levels of accuracy when detecting mosaic variants when compared to detecting somatic mutations, especially in the case of cancer, despite using deep-detection methods. Improvements need to be made in this domain.
One of the most important components to have when benchmarking variants is to construct a reference standard that can be used whenever required by the user in a convenient manner. Some popular examples of such standards are Genome in a Bottle and CRISPR-Cas9. Genome in a Bottle is relevant for germline mutations, and CRISPR-Cas9 is a genome editing tool that has been used for inserting mutations to produce products for commercial use. BAM-File mixing is a simple yet efficient method of generating simulated datasets in silico. A significant advantage offered by them is that it generates a very simple error profile and reduces unnecessary noise, such as redundant sequencing errors. It is also one of the researchers’ most strongly suggested methods for mosaic variant analysis. Unique molecular identifiers (UMI) and linked-red sequencing have been found to be effective for detecting variants with low VAF scores (less than 5%). Further developments have to be done to improve their accuracy.
Limitations and Future Potential
Some of the most pronounced limitations of existing methods for mosaic variant detection are the limited search space, dependency on data, and discrepancies in the results obtained after conducting analysis on the genome under study. A few confusions also need to be cleared; it is important to ensure that strategies used for mosaic calling are not confused with baseline algorithms. These strategies are simply modified versions of the original algorithms, and more development is needed to test whether they could be potentially useful when specific algorithms are unavailable.
Benchmarking is largely limited due to complex parameters used in this study – as of now, default parameters are preferred due to this reason. Deep exome sequencing is based on capturing reads across the entire exon region in depth. While the accuracy of this technique is good, it still shows decreased levels of detecting low VAFs, as the coverage is across an uneven space. Regions of low complexity, such as repeats and duplications within segments, were excluded during the benchmarking process, even though they are an area of research that many developers and researchers are pursuing. This was done due to the existing usage of filtration strategies in such regions, and researchers wanted to test the abilities of this technology in more core regions. Any shifts in the reference standards composition can cause alterations in results, such as read length, the distributions of datasets containing VAFs, and positive controls present in the study.
Conclusion
This study aims to guide researchers to help them choose the algorithms suited to their needs, as well as for developers to build models that can aid in studying mosaic variants present in somatic cell lines and germlines. Understanding mosaic variants better will gradually help us understand fundamental processes that are linked to the development of living organisms.
Article source: Reference Paper
Learn More:




Swasti is a scientific writing intern at CBIRT with a passion for research and development. She is pursuing BTech in Biotechnology from Vellore Institute of Technology, Vellore. Her interests deeply lie in exploring the rapidly growing and integrated sectors of bioinformatics, cancer informatics, and computational biology, with a special emphasis on cancer biology and immunological studies. She aims to introduce and invest the readers of her articles to the exciting developments bioinformatics has to offer in biological research today.











{kind=link}