
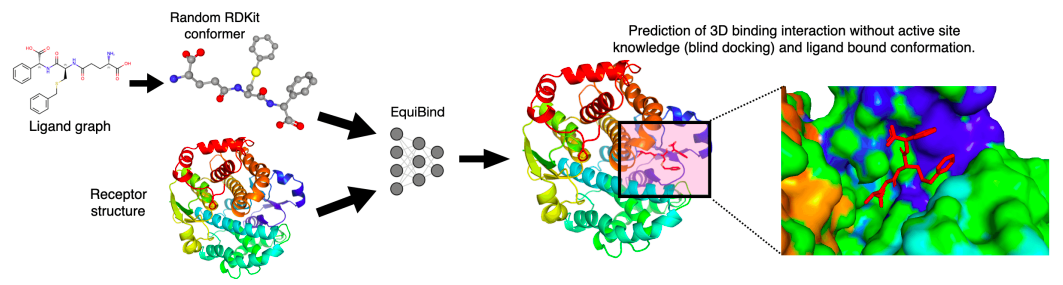
MIT researchers have developed a geometric deep-learning model, EquiBind, which is 1,200 times faster than one of the fastest computational molecular docking models currently in use, QuickVina2-W, for effective binding of drug-like substances to proteins.
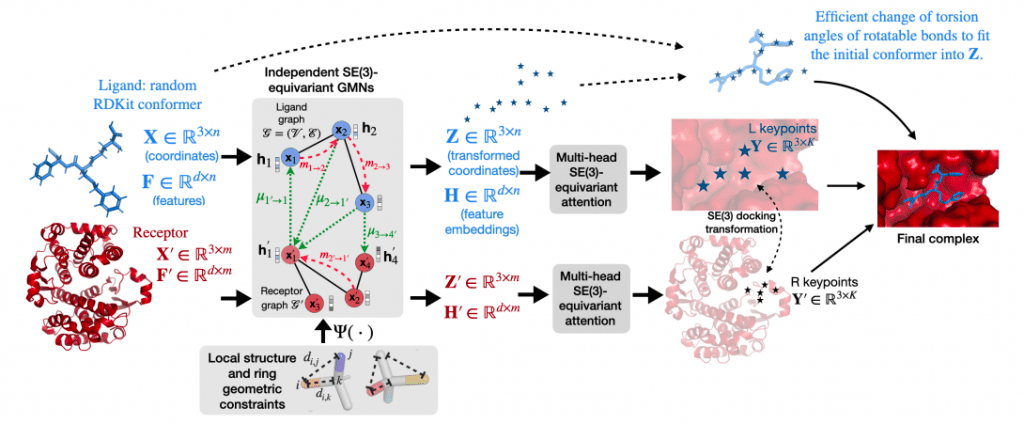
Image Source: https://arxiv.org/pdf/2202.05146.pdf
One of the main challenges in drug discovery is to predict the binding properties of a drug-like chemical to a certain protein target.
Key applications like quick virtual screening or drug engineering would be made possible by an incredibly quick computational binding approach.
Existing techniques require a lot of candidate sampling along with scoring, ranking, fine-tuning stages, which makes them computationally expensive.
With EquiBind, a SE(3)-equivariant geometric deep learning model that performs direct-shot prediction of the receptor’s binding location (blind docking) as well as the ligand’s bound pose and orientation, the scientists challenged this paradigm.
Comparing EquiBind to conventional and contemporary baselines, it achieves notable speed-ups and better quality.
Furthermore, at the expense of longer run times, the researchers have demonstrated additional improvements when coupling it with current fine-tuning methods.
Finally, in order to avoid the previous costly differential evolution strategies for energy minimization, the researchers have suggested a novel and quick fine-tuning model that modifies the torsion angles of a ligand’s rotatable bonds based on closed-form global minima of the von Mises angular distance to a given input atomic point cloud.
Major Issue in Drug Discovery
Understanding how ligands interact and form complexes with target proteins or receptors is a key challenge in drug discovery and a requirement for virtual screening.
This is a challenging topic with many features and restrictions. The domain knowledge characterizing ligand-protein binding mechanisms includes binding kinetics, conformational changes (internal molecular flexibility), and diverse forms of chemical and geometrical atomic interactions.
For instance, “lock-and-key,” “induced fit,” and “conformational selection” are conventional models for the formation of molecular complexes, but the most frequent atomic interactions during binding are hydrophobic, hydrogen-bonding, and π-stacking.
EquiBind
In the given study, the scientists presented EquiBind, a brand-new geometric and graph deep learning model for structural drug binding.
The researchers used graph matching networks (GMN) and E(3)-equivariant graph neural networks (E(3)-GNN) to perform a direct prediction of the structure of the ligand-receptor complex without relying on heavy sampling as in previous work, resulting in significant inference time speed-ups.
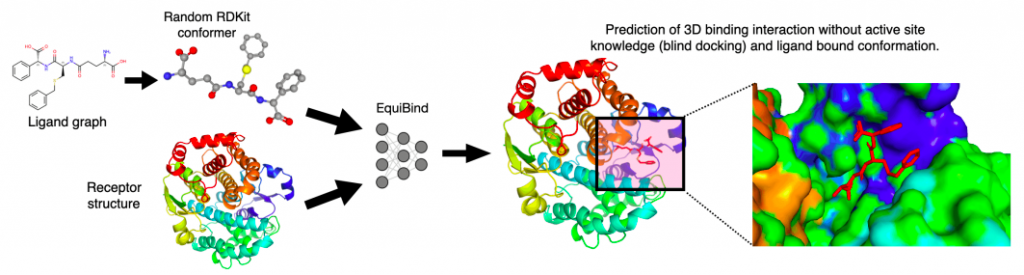
Image Source: https://arxiv.org/pdf/2202.05146.pdf
Injecting the appropriate physical, chemical, or biological inductive biases into DL models is also essential because 3D structural data is scarce (for example, only about 19K experimental complexes are publicly available in the PDBbind database).
By doing this, one can avoid learning from insufficient amount of data and produce reliable models.
In order to achieve this, EquiBind:
- Ensures independence to the initial 3D placements and orientations of the two molecules, i.e., the same complex is always predicted for the same input unbound structures;
- Incorporates an effective mechanism for biologically plausible ligand flexibility by only altering torsion angles of rotatable bonds while keeping local structures (bond angles and lengths) fixed; and
- Utilizes a non-intersection loss to prevent steric clashes or interference.
Demonstrations from Experimental Analyses
The scientists concentrated on the blind molecular docking scenario, which involved having no knowledge of the binding location or pocket of the protein.
However, this approach was readily applicable where the approximate binding position is known. The scientists contended that mistakes in the ground truth binding pocket conformation have a significant impact on conventional molecular docking techniques that depend on the receptor active site.
In reality, the ground truth 3D positions of the binding atoms may be low-resolution or unknown at all (for example, for novel antigens), or the researchers may have been interested in finding new druggable sites on the surface of a protein that was previously believed to be undruggable (e.g., exploring allosteric binding locations rather than orthosteric sites).
Empirically, the researchers looked into two situations: flexible self-docking and redocking, which involved removing the bound ligand structure from a complex and asking the model to dock it (i.e., ligands have no bound structure knowledge prior to molecular docking).
Assuming a rigid receptor, the researchers described the flexibility of the ligand by first predicting an atomic point cloud of the bent molecule and then using a quick technique to extract internal changes in the torsion angles of rotatable bonds that would closely match the point cloud.
The researchers maximized the log-likelihood of a von Mises distribution that fitted the torsion angles, proving closed-form expressions of the global optimum instead of minimizing the root-mean-square deviation (RMSD) using pricey optimization techniques (e.g., differential evolution approaches).
Using several criteria, the scientists experimentally demonstrated enhanced quality over current and popular best-in-class baselines at a small fraction of the running time.
Finally, the researchers demonstrated the efficacy of utilizing EquiBind in conjunction with current energy-based techniques to produce a hybrid DL approach. The researchers thought that the paradigm illustrated in the study would be used in computational drug discovery in the future.
The Endpoint
Deep neural networks‘ potential to predict protein structures has generated many studies in the field of computational drug discovery. In this study, the scientists put forth the EquiBind deep neural model, which predicts bound protein-ligand conformations in a single pass using SE(3)-equivariant graph neural networks.
By merging their model with current fine-tuning techniques, the researchers demonstrated its potential in a hybrid workflow and showed that it performs well empirically against best-in-class baselines. The researchers anticipated that the use of deep learning in drug development would advance thanks to models like EquiBind and others.
Article Source: Stärk, H., Ganea, O., Pattanaik, L., Barzilay, R., & Jaakkola, T. (2022, June). Equibind: Geometric deep learning for drug binding structure prediction. arXiv. Preprint. https://arxiv.org/pdf/2202.05146.pdf
Learn More About Bioinformatics:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}