
Proteins perform essential functions in living organisms and adopt specific three-dimensional structures dictated by their amino acid sequences. The ability to accurately design novel protein structures from scratch could enable advances in areas like protein engineering and drug development. Recently, deep learning techniques like diffusion models have shown promise for generating designable protein structures without predefined templates. This article explores a new method called Proteus that pushes the boundaries of protein structure generation through innovations in model architecture and training strategies.
De novo protein design aims to create novel protein folds that can be realized by at least one amino acid sequence. Accurate structure prediction models have opened up opportunities for generating designable proteins by using the models in reverse. RFDiffusion demonstrated this by fine-tuning a structure predictor called RoseTTAFold and achieving high experimental success rates. However, reliance on pre-training makes further model development difficult. Other diffusion models, such as FoldingDiff and Genie, avoid pre-training but lag far behind in designability.
To address these limitations, the researchers from Westlake University, China, introduced Proteus – a novel protein structure diffusion model that rivals RFDiffusion’s designability without pre-trained weights. Proteus combines a multi-track architecture, graph-based triangle attention, and improved training strategies to achieve state-of-the-art performance. In computational evaluations and lab experiments, it generated designable structures with remarkable efficiency.
Protein Structure Representation
Proteins are represented by a backbone “frame” for each amino acid residue, encapsulating the 3D positions and orientations. These frames are rigid transformations in the special Euclidean group SE(3), comprising a rotation matrix R ∈ SO(3) and translation vector t ∈ R3. R is derived from the backbone atoms N, Cα, C, and t, which corresponds to the Cα atom position.
Diffusion Modeling
Diffusion models are trained to predict improved “denoised” data x0 given corrupted data xT from some noise distribution. For protein backbones, forward diffusion is modeled as Brownian motion on SO(3) and R3. By estimating the probability distribution, the score matching loss trains a model fθ to predict ∇xT log pT(xT|x0) and can be used to reverse the diffusion process.
Relevant Network Architectures
Alphafold2’s Evoformer translates sequence and alignment data into per-residue representations. Its Structure Module then iteratively refines an initial structure prediction. RoseTTAFold uses a similar architecture. The triangle attention mechanism in Evoformer is important for modeling structural constraints but is highly complex.
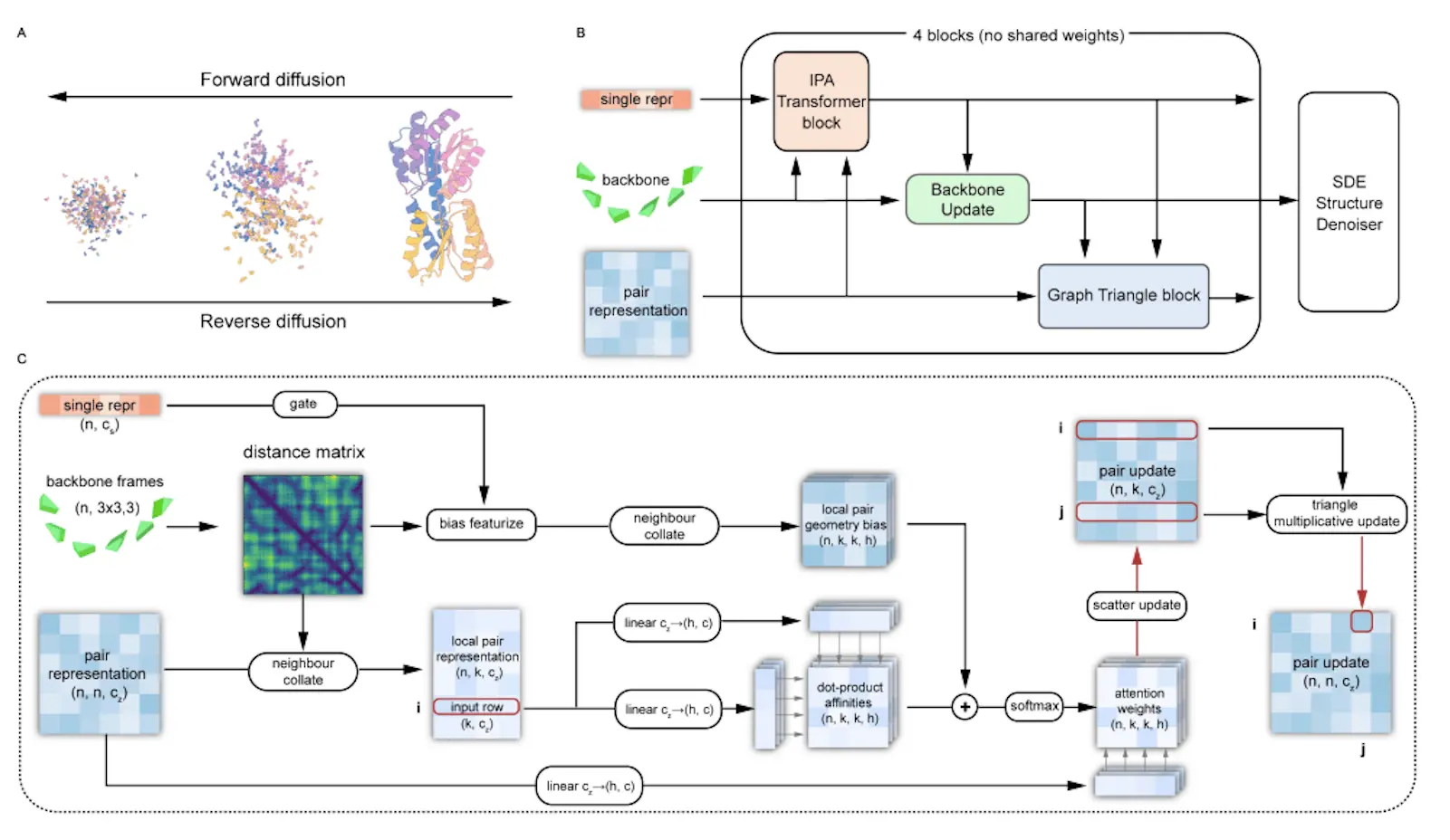
Proteus comprises a series of folding blocks with multi-track inputs: node features, edge features, and coordinate frames. The core innovations are:
IPA-Transformer Block: Uses IPA attention biased by distance and edge features, plus a standard Transformer layer. Enhances representation learning.
Backbone Update: Adapted from Alphafold2 to predict coordinate frame updates.
Graph Triangle Attention: Novel block that applies triangle attention to a graph of K nearest neighbor residues instead of full O(N^3) complexity. This significantly enhances edge features by incorporating structural bias and multi-track interactions.
Proteus is trained without pre-training by optimizing the weighted combination of a denoising score matching loss and auxiliary coordinate prediction losses. The training dataset was derived from PDB by splitting complexes into single chains and retaining high-quality monomers up to 512 residues.
Proteus Performance
Monomer Generation:
Proteus achieves state-of-the-art designability across different monomer lengths, even exceeding RFDiffusion. For longer sequences ≥400 residues, the performance gap widens further. Proteus also matches RFDiffusion in diversity and Chroma in efficiency. The “success efficiency” metric combining designability and speed highlights Proteus’ advantages.
Complex Generation:
Proteus substantially outperforms Chroma at generating designable dimers, trimers, and tetramers. This demonstrates its potential for multi-domain proteins and nanoparticles.
Ablation Studies:
Graph triangle attention and single-chain data augmentation contribute significantly to Proteus’ designability gains. The rotational self-conditioning approach from Alphafold2 also provides noticeable improvements.
Experimental Validation:
SEC, CD spectroscopy, and temperature ramping verified that 9/12 300-residue and 3/4 500-residue Proteus designs expressed as stable, folded proteins.
Proteus: Redefining Protein Structure Generation
Proteus establishes a new state-of-the-art for unsupervised protein structure generation models. By innovating graph-based triangle attention and multi-track communication, it exceeds even the pre-trained RFDiffusion in designability and efficiency. This performance edge originates from:
- Enhanced representation learning and constraint modeling with triangle attention
- Reduced complexity via graph computation
- Improved generalizability from augmented training data
- Faster high-quality sampling requiring fewer diffusion steps
Proteus also showed promising results on complexes and experimental validation. Its advanced generative capabilities could enable progress in protein design, co-folding for drug discovery, and nanoparticle engineering.
Future work involves extending Proteus to model sidechains and ligands for complete co-folding and integrating sequence generation networks for end-to-end protein design. As shown by Alphafold2’s open-source release, putting enhanced generative models in the hands of researchers can accelerate innovation. By similarly open-sourcing Proteus, the researchers aim to supercharge efforts in computational molecular design.
Conclusion
This article covered Proteus, an unsupervised protein structure diffusion model that achieves new heights in designability and efficiency. Proteus advances the state-of-the-art backbone generation without reliance on pre-training by reformulating triangle attention with graph computation and multi-track communication. Its performance could enable more rapid and effective protein design, expanded co-folding capabilities, and new frontiers in nanomaterial engineering. By combining innovations in neural architecture with a commitment to open science, this work exemplifies the transformative potential of AI in computational molecular design.
Article source: Reference Paper
Follow Us!
Learn More:



Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











{kind=link}