
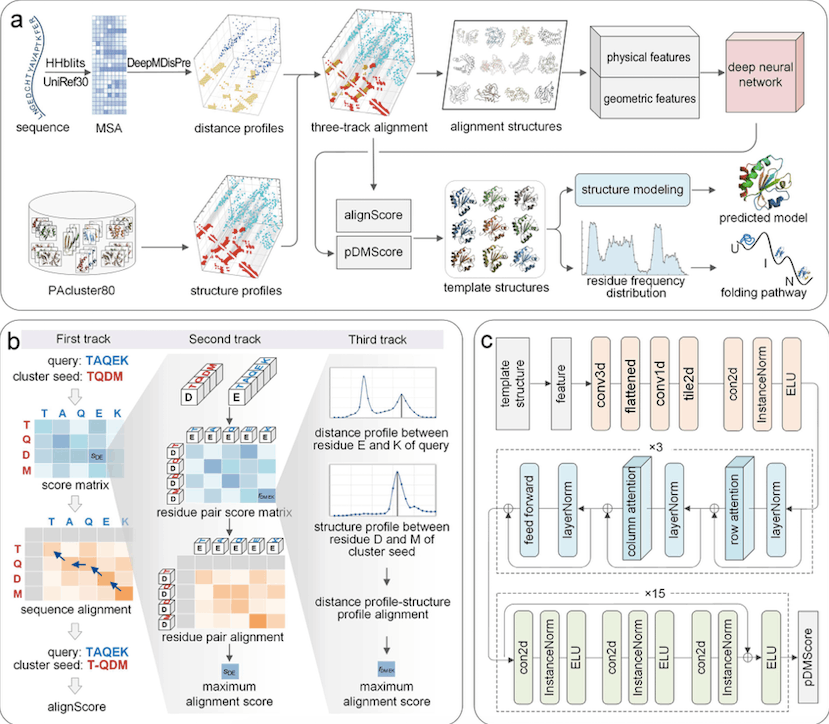
Scientists from the Zhejiang University of Technology, China, have proposed a new method, PAthreader, to improve the recognition accuracy of remote homologous structures by designing a three-track alignment between predicted distance profiles and structure profiles extracted from the PDB and AlphaFoldDB. As a result, AlphaFold2 performs better, and structural modeling is more precise. The results demonstrate the possibility of utilizing folding information from distant homologous structures by showing positive predictions for 37 proteins, including seven that align with biological investigations.
Protein Structure Modelling: Template, Methods, and Limitations
The DeepMind team has made a significant advancement in machine learning-based protein structure modeling with their AlphaFold2 algorithm. However, AlphaFold2 does not investigate the mechanics of how proteins dynamically fold into their equilibrium structures. It is well known that templates play an important role in the modeling of protein structures. Templates are crucial for enhancing the precision of protein structure prediction. In general, protein structure modeling techniques fall into three categories: physics-based methods, knowledge-based methods, and end-to-end deep learning methods.
Attempts to replicate the folding process with equilibrium MD simulations and other approaches have been made. The evolutionary connections across protein families may carry implicit information about the folding of individual proteins. In the present research, the most prevalent template recognition methods may be loosely classified into two categories: profile-based alignment methods and binary contact/distance-based threading methods.
Working of PAthreader
PAthreader is a new technology presented by researchers for detecting distant templates and studying the protein folding route. They trained their algorithm using Protein Data Bank (PDB) and AlphaFold database information. PAthreader outperformed other state-of-the-art approaches for remote template recognition, according to the findings. In addition, they studied the interaction between the template and the AlphaFold2 model and discovered that the model’s quality was contingent on the availability of a decent template. Using PAthreader to detect folding intermediates from homologous templates, they were able to derive protein folding pathways for seven instances and thirty human proteins. The authors also hypothesized that the evolutionary link between protein families implicitly contains information about the folding of individual proteins.
PAthreader is a method for identifying protein folding routes and detecting distant templates. It uses DeepMDisPre to obtain multi-peak distance profiles and PAcluster80 database structure profiles. The query sequence is aligned to each cluster seed of PAcluster80 using a three-track alignment technique, and physical and geometric properties are extracted and input into a convolutional network to predict the DMScore. The best templates are subsequently used for structural modeling, and the folding route is explored based on folding intermediates.
Comparison with other Models in Protein Structure Prediction: SCOPe37 and AlphaFold Database
PAthreader’s performance was compared to those of other state-of-the-art methods, such as HHsearch and LOMETS3. The research used a test set of 551 proteins from the SCOPe37 database with sequence identities of less than 30%. The lengths of the proteins ranged from 120 to 700 residues, and their resolution was less than 2.
The test set was subdivided into four subgroups depending on the TM-score of the best template in the Protein Data Bank in order to examine the effectiveness of various approaches at varying difficulty levels (PDB). A TM-score of 1 indicates a perfect match between two protein structures.
PAthreader outperformed HHsearch and LOMETS3, as the average TM-score of the test targets was 12.2% and 5.2% higher for PAthreader than for HHsearch and LOMETS3, respectively. The P-values of the Student’s t-test demonstrated that there was a statistically significant difference between PAthreader and the other approaches.
When the test set was categorized based on the number of domains in the proteins, PAthreader was shown to be superior to HHsearch and LOMETS3 for template recognition of both single-domain and multi-domain proteins.
AlphaFold DB, which improves the family coverage of model organism proteomes and includes unique folding structures and motifs not present in PDB, is one of the reasons for PAthreader’s superior performance. Ablation studies were undertaken to determine the influence of AlphaFold DB on template recognition, and it was shown that the performance of the PAthreader improved significantly when AlphaFold DB was applied to the more challenging targets.
Analyzing the Relationship between the Accuracy of AlphaFold2 Predictions and the Quality of Templates
Two studies were conducted to determine the effect of template quality on AlphaFold2’s performance. The first experiment evaluated the quality of templates for high-scoring models in the AlphaFold Database, which contains over 500,000 protein structures from 48 organisms. More than half of the high-scoring models in AlphaFold DB have templates with a TMscore of at least 0.8 in the Protein Data Bank (PDB), demonstrating that AlphaFold2’s success is contingent upon the quality of the template.
In the second experiment, the scientists examined the link between the accuracy of the model and the initial template used for AlphaFold2 modeling on 551 test proteins. They discovered that models with a TMscore of at least 0.9 were developed for 88% of the targets with higher quality templates (TM-score 0.7), however, the number reduced to 49.8% for targets with relatively poor templates (TM-score 0.7). In rare circumstances without high-quality templates, AlphaFold2 can nonetheless produce high-scoring models, either due to the utilization of plentiful multiple sequence alignments (MSAs) or because the test proteins were included in AlphaFold2’s training set.
The results indicate that the performance of AlphaFold2 depends to some extent on the quality of the template and that giving higher-quality templates may increase its performance.
Comparison with other Models in Protein Structure Prediction: Template Recognition and TM-Score Analysis
PAthreader developed better models with higher TM scores than HHsearch in the majority of situations, as determined by an analysis of the association between model accuracy and the initial template used for PAthreader modeling on CAMEO targets. Also, the study discovered that PAthreader obtains templates with TM-scores 0.90 on nearly twice as many targets than HHsearch.
PAthreader generated better models than AlphaFold2 and PureAF2_orig by discovering better templates with higher TM-scores. The study gave two typical examples of a single-domain protein and a multi-domain protein.
PAthreader was used to recognize templates and model structures for 17 SARS-CoV-2 viral proteins, producing a slightly better average TM-score than AlphaFold2 models. PAthreader’s model got a higher TM-score than AlphaFold2’s model, according to a comparison of their structural models.
Predicting Protein Folding Pathways
PAthreader predicts protein folding routes by studying distant homologous protein structures. The method involves matching the templates with the target protein, segmenting it based on its secondary structure, and calculating ResFscore to find folding intermediates. The ResFscore distribution of identified residues from homologous templates can provide an explanation for the folding order revealed by biological tests. The method was used for seven instances and thirty human proteins, including horse heart cytochrome c, and the anticipated folding mechanism was compared to experimental findings. The results indicate that intermediates are present in the selected template structures.
Conclusion
PAthreader is a remote template recognition and protein structure prediction application. Due to its extended model organism proteomes, excellent sequence-template alignment, and physiologically reasonable template selection based on anticipated pDMScore, it outperforms other approaches in finding distant templates. With 10 Processors, the software can identify templates for tiny proteins in half an hour. PAthreader was used to enhance AlphaFold2, which increased its precision in predicting multi-domain proteins and was rated first in the CAMEO blind test for three months. Nevertheless, upgrading templates can only enhance AlphaFold2 to a limited level, and incorporating PAthreader into AlphaFold2 and retraining it would need a large number of computing resources.
Article Source: Reference Paper | PAthreader: Webserver
Learn More:




Sejal is a consulting scientific writing intern at CBIRT. She is an undergraduate student of the Department of Biotechnology at the Indian Institute of Technology, Kharagpur. She is an avid reader, and her logical and analytical skills are an asset to any research organization.





{kind=link}