
Scientists from China have developed DeepBIO, an automated and interpretable deep learning platform for high-throughput biological sequence functional analysis. The first-of-its-kind platform enables researchers to develop new deep-learning architectures addressing particular biological questions. For any given biological sequence data, DeepBIO offers 42 state-of-the-art deep learning algorithms for model training, comparison, optimization, and evaluation in a fully automated pipeline. The pipeline enables ultra-fast predictions and enormous improvement in computational speed with up to million-scale sequence data within a few hours with the aid of high-performance computing and GPUs. The authors envision that DeepBIO will ensure the reproducibility of deep-learning sequence analysis and provide meaningful functional insights from sequences alone.
The precursors to DeepBIO and why we need it
The current next-generation sequencing techniques generate vast amounts of sequence data that require fast, functional analysis tools. The large-scale data ranging from genomic and transcriptomic to proteomic sequences poses great challenges for wet laboratory functional analysis experiments. Machine learning-based methods have eased the understanding of complex mapping from biological sequences to their structures and functional mechanisms. Several machine learning-based methods have been developed over the last decade to study functional aspects of biological sequences. BioSeq-Analysis was the first platform developed for analyzing various biological sequences using machine learning. BioSeq-Analysis 2.0 was later developed for the automatic generation of various predictors for sequence analysis both at the residues and sequence levels.
The platform iLernplus provides a pipeline for sequence analysis which consists of feature extraction and selection and model construction as well as prediction results analysis. BioSeq-BLM is a web platform that provides different language models for DNA, RNA, and protein sequence analysis. Other such tools include iFeatureOmega, Rcpi, and ProtrWeb.
All the above methods require strong professional knowledge for training the models, and this reduces the user base drastically. Deep learning has proved to be a reliable alternative to machine learning-based tools due to its scalability as well as adaptivity. Deep learning-based tools for functional analysis of biological sequences have been developed and include Kipoi, Pysster, and Selene. While these methods use the deep learning-based method development advantages to some extent, however, they are not completely automated and require computer science skills and knowledge to be able to set up the method and use it. Thus the need for an automated deep learning platform for functional analysis of biological sequences. With this in mind, the authors developed DeepBIO.
The DeepBIO platform framework
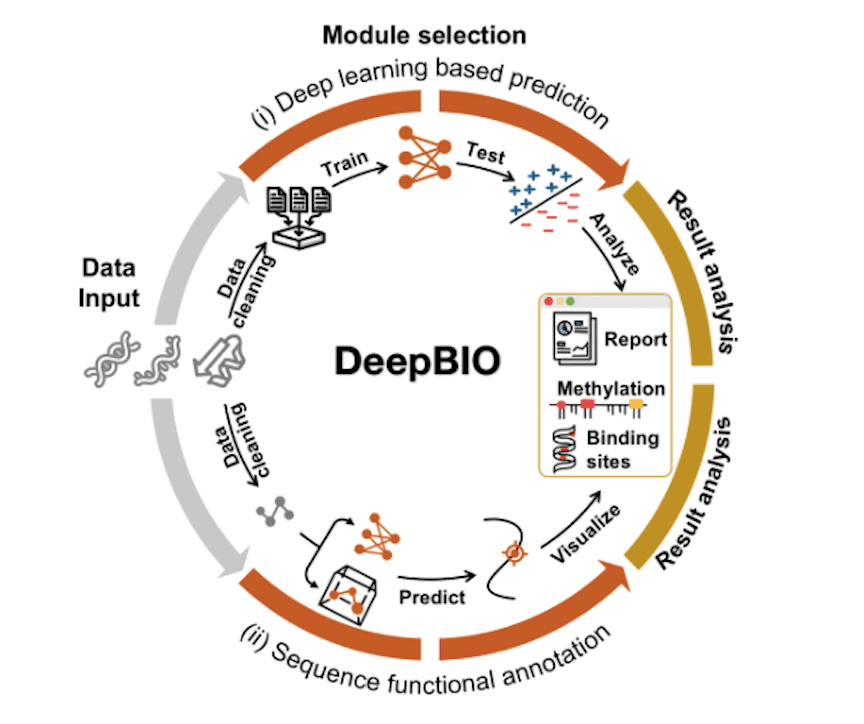
The authors developed DeepBIO, a web platform for sequence analysis, functional annotation, and result visualization using an automated deep learning-based pipeline. It consists of four modules: a data input module, sequence-level functional prediction module, base-level functional annotation module, and a result report module. The input module can handle DNA, RNA as well as protein sequences. The second module enables users to automatically train, evaluate, and compare the deep learning models with their input data. The third level provides predictions of DNA methylation, RNA methylation, and protein binding specificity. The last module generates visualization analysis results in different data formats. The following figure illustrates the framework components in detail.
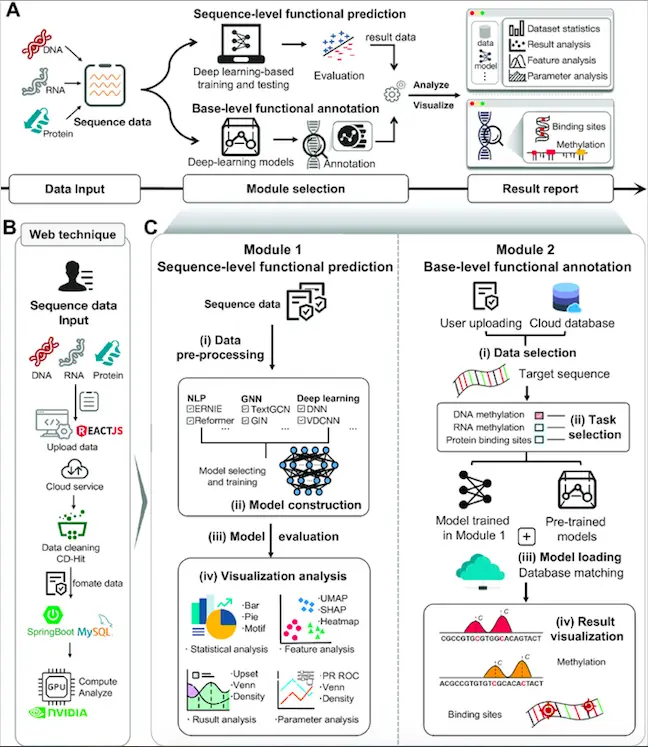
Image source: https://doi.org/10.1093/nar/gkad055
Applying DeepBIO to real-world data
The authors apply the automated pipeline to real data under two scenarios. First, the prediction of DNA 6mA methylation and second, the prediction of protein toxicity. DNA 6mA methylation is involved in genomic imprinting as well as carcinogenesis. On the other hand, protein toxicity is the underlying cause of many neurodegenerative diseases. The case studies illustrate the span of usability of the pipeline for both fundamental research as well as for therapeutics.
For the first task, the Drosophila melanogaster dataset with 11,191 methylation sequences was used. The second case study involved 3,379 toxic animal protein sequences from a benchmark dataset.
The authors chose the DNABERT, DNN, LSTM, and RNN deep learning models to train and compare their prediction performance on the first dataset. This illustrates that the platform offers more than forty deep-learning models to choose from for training and comparison with the input data. The authors also illustrate the feature analysis and visualization aspects of the platform for the datasets as well as the functional annotation analysis. The following figure illustrates the prediction analysis results from the different models used.
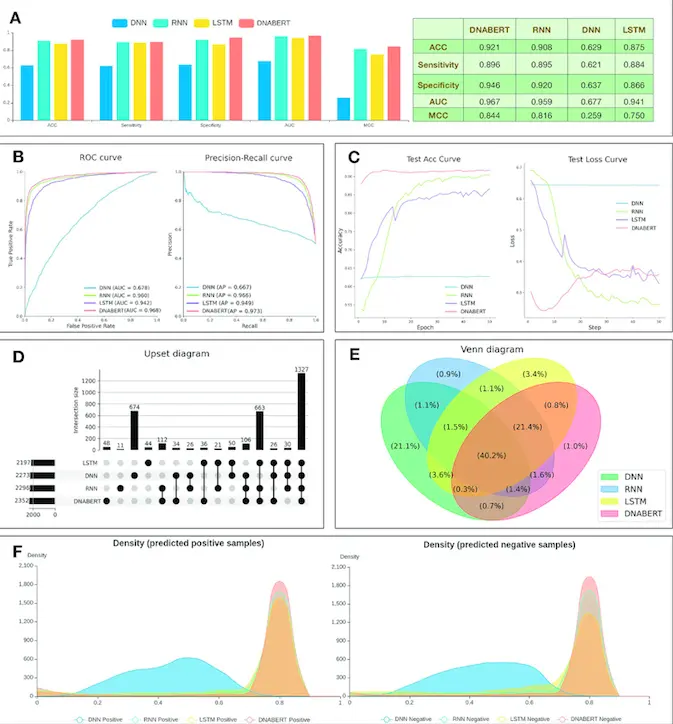
Image source: https://doi.org/10.1093/nar/gkad055
Ultra-fast performance
DeepBIO, aided by high-performance computing, is capable of performing calculations at the scale of millions in a few hours. This is achieved by using GPUs for training and model predictions, unlike other platforms that use CPUs instead. The authors have shown that DeepBIO spent less time optimizing deep learning models as compared to others. DeepBIO has also proven to be fast and accurate in its predictions.
Conclusion
With the advent of deep learning-based approaches in analyzing biological sequences, the need for a fast, accurate and completely automated, and interpretable platform was ever-rising. The authors successfully developed and present DeepBIO, the web platform for biological sequence analysis, functional annotation, as well as visualization analysis, all under the same umbrella. This is indeed a breakthrough in bioinformatics, as the platform generalizes over input data and allows the user to select from more than forty deep learning models to train and compare their input data with. Backed by high-performance computing and the use of GPU clusters for training and generating predictions, the pipeline promises to be a significant tool to test for the reproducibility of deep learning-based results and promises to aid both fundamental research as well as drug design and delivery.
Article Source: Reference Paper | DeepBIO: Web Platform
Learn More:




Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





{kind=link}