
A team of researchers led by Prof. Jiangning Song from Monash University, Australia, has developed a novel ML ensemble predictor named PreAcrs that can identify anti-CRISPR proteins from a given polypeptide sequence with good accuracy and robustness.
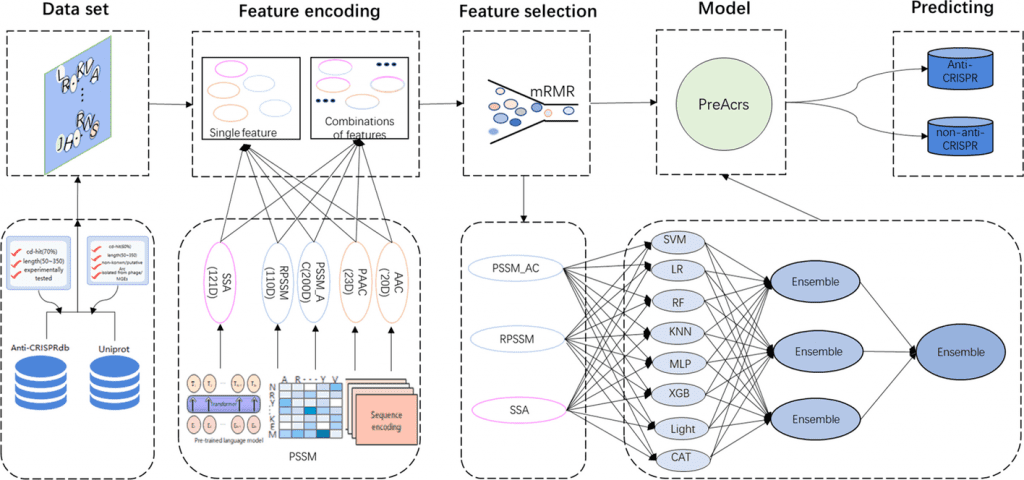
Image Source: https://doi.org/10.1186/s12859-022-04986-3
Clustered regularly interspaced short palindromic repeats (CRISPR) is a family of DNA sequences, CRISPR-associated protein (Cas) is a family of endonucleases ( an enzyme that can cleave specific strands of DNA) commonly found in the genomes of bacteria and archaea. The CRISPR-Cas together act as an adaptive immune system protecting the prokaryotes against invading nucleic acids. The life sciences domain has been completely transformed by CRISPR, the innovation that won the 2020 Nobel Prize in Chemistry. This approach could revolutionize the treatment of genetic diseases, cancer, and other conditions by making precise genome alterations quickly, cheaply, and efficiently.
Found serendipitously by a doctoral student at the University of Toronto, Anti-CRISPR proteins are small proteins (50-150 amino acids in length) generated by bacteriophages that suppress the CRISPR Cas system in bacteria. Although there are several approaches, such as the,
1) Guilt-by-association bioinformatic analysis – looks for homologs of proteins with helix-turn-helix (HTH), often encoded after anti-CRISPR proteins.
2) Self-targeting CRISPR arrays
3) Metagenome DNA screening
These models work by assuming a new anti-CRISPR protein might be similar to old, well-studied anti-CRISPR proteins. But they fall short because of unreliability and lack of extensive knowledge of anti-CRISPR proteins.
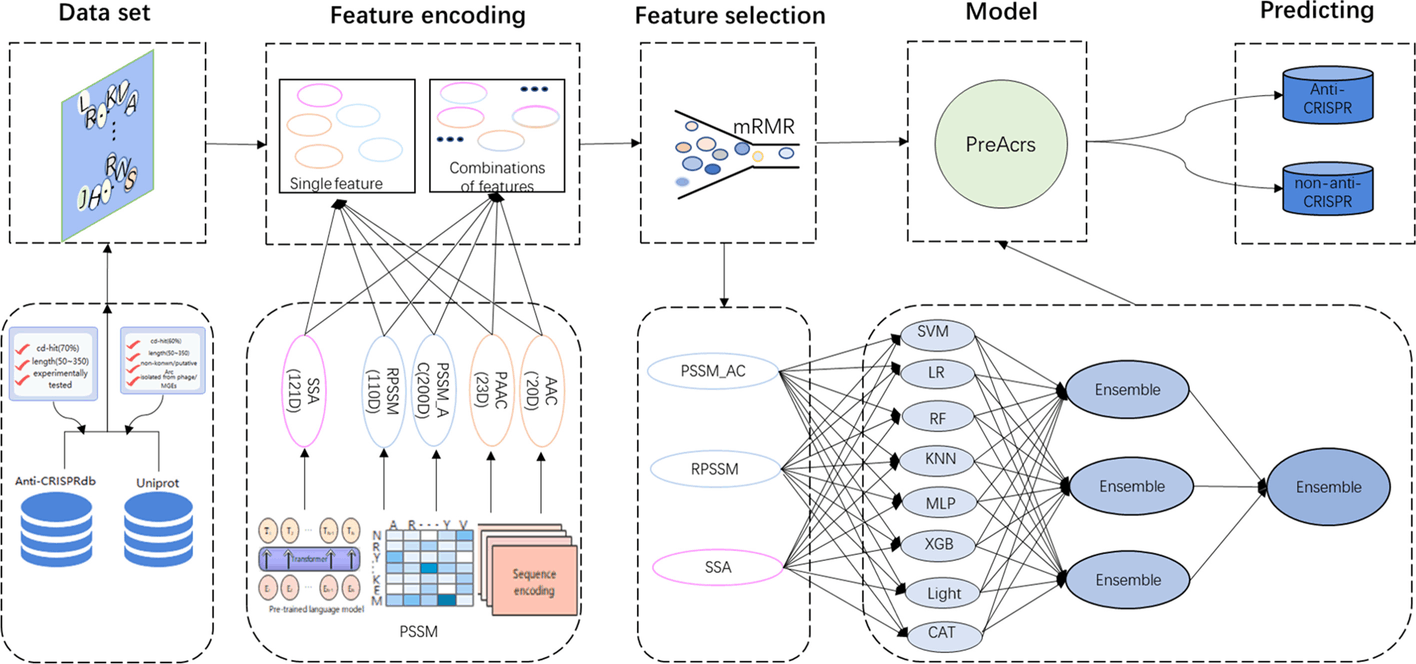
Several machine learning web servers such as Anti-CRISPRdb, AcrHub, AcrDB, CRISPRminer2, AcRanker, AcrFinder, ArcCatalog, and PaCRISPR have been proposed to identify anti-CRISPR proteins from a given protein sequence, but they fail to assess and identify anti-CRISPR proteins in a holistic view. Thus, PreAcrs was created. The model was trained using an extensive array of three unique features and eight different machine-learning methods.
Major Findings
The authors introduced 412 experimentally validated and 412 non-anti-CRISPR proteins followed by an independent dataset consisting of 176 experimentally determined and 176 non-anti-CRISPR proteins were also considered. Three features, RPSSM, PAAC AC, and SSA, out of a total of five feature encoding methods (AAC, PAAC, PSSM AC, RPSSM, and SSA), were taken into consideration for their accuracy. With an AUC of 0.967 and 0.961, two ensemble features—PSSM AC & RPSSM and RPSSM & SSA—achieved good performance. As a result, the feature RPSSM contributed the most to the PreAcrs model’s ability to forecast anti-CRISPR proteins (Acrs). Additionally, since single features cannot fully capture the Acrs for identification, the authors made two attempts to combine five single characteristics: ensemble features and combination features. In order to train models, combination features combine individual components into a vector. With the highest AUC value, the ensemble feature PSSM AC & RPSSM & SSA demonstrated the best performance. PAAC & PSSM, AC & RPSSM is the second-best, and PSSM AC & RPSSM is the third-best ensemble feature.
We contrasted PreAcrs with the most advanced Acrs predictor PaCRISPR in order to further assess the efficacy of the PreAcrs predictor. The PreAcrs approach is superior to other predictors for capturing the inherent patterns of non-homologous Acrs. PaCRISPR only took into account four features, whereas PreAcrs used eight different models, including the SSA feature from the pre-trained model, signifying its robustness and prominence to other Acrs predictors.
Final Remarks
The ability to regulate CRISPR-Cas machinery as a tool for gene editing or gene therapy strongly relies on identifying potential Acrs. It is possible to discover Acrs more quickly using the machine learning method based on the protein sequence. PreAcrs, an ensemble framework based on machine learning, could precisely and successfully detect Acrs from protein sequences by combining the evolutionary features with the pre-trained model feature. Although PreAcrs performs very well, it still has some drawbacks. One drawback is that the mRMR algorithm is used to choose significant features in PreAcrs, which could lead to some biases that lower the forecast accuracy. PreAcrs’ website does not provide a user-friendly platform, making it challenging for biologists to evaluate Acrs. Therefore, working towards a potent, user-friendly, and interactive website that may utilize several feature selection algorithms can be a way forward.
Article Source: Reference Paper | Code and Data Availability: GitHub
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Shwetha is a consulting scientific content writing intern at CBIRT. She has completed her Master’s in biotechnology at the Indian Institute of Technology, Hyderabad, with nearly two years of research experience in cellular biology and cell signaling. She is passionate about science communication, cancer biology, and everything that strikes her curiosity!





{kind=link}