
Stanford University researchers have created a graph-based algorithm for reconstructing interactomes and identifying clinically important cell-cell interactions. The algorithm can be used to create a global network of cell-to-cell cross-talk.
REMI is a modernized, novel algorithm that utilizes graph-based ways to deal with creating a global cell-cell interaction network by assessing the conditional dependency of LR pairs on high-dimensional data of a small sample size. Instead of current strategies, REMI catches the intricacy of multicellular interactions by representing the potential confounding impacts that LR pairs may have upon each other.
Cell-cell interactions between and inside the different cell types containing the tissue microenvironment assume an essential part in controlling local and fundamental biological and physiological capacities under typical and neurotic circumstances. These connections work with collaboration or contest between cell types and are commonly mediated among ligands and receptors. Ligands are frequently shown as dissolvable or extracellular proteins expressed by the “sending” cells and tie onto a related receptor on the “getting” cells. In tumor microenvironments (TMEs), cell cross-talk between tumor, stroma, and immune cells arranges the foundation of preinvasive and invasive niches that empower cancer encouraging properties, like tumor development, immune evasion, and metastasis.
Enormous scope cellular interactions are challenging to gauge utilizing mutt lease trial methods, yet a few computational methodologies have been proposed to foresee these communications using – omics information. A greater part of the current computational methodologies that utilize high-throughput transcriptomics information to surmise cell-cell interactions either ascertain interaction scores based on quality articulation stage tests or execute chart-based approaches. In expression change based approaches, for example, CellphoneDB v2.0, what’s more, NATMI, possible ligand and receptor (LR) interactions are recognized by thresholding the qualities based on their expression level with the supposition that this predicts higher LR protein abundance.
Other different strategies, like CCCExplorer and NicheNet, compute correlation metrics between the expression levels of the ligand, receptor, or downstream signaling pathway genes for each LR pair.
Notwithstanding, a correlation between the expression of ligand, receptor, or downstream genes might be catching an indirect association brought about by another LR interaction, including the ligand or, on the other hand, a receptor of interest.
While current crosstalk surmising approaches give a valuable pattern for incorporating hypotheses of LR interactions, they do not catch the conditional dependencies of LR pairs among multiple cell types. Current methodologies assume that pairwise interactions are autonomous, yet pairwise interactions can be affected by different interactions utilizing autocrine and paracrine loops.
Computing the conditional dependencies of LR pairs is a poorly characterized issue for high-dimensional datasets created across a small sample size. This challenge is normal in omics data analysis due to the huge number of parameters (p) and the relatively small number of samples (n). An algorithm called REMI (REgularized Microenvironment Interactome) was presented to recognize communities of dependent LR pairs in high-dimensional datasets of small sample sizes utilizing graph-based approaches to address this challenge.
The presentation of REMI was exhibited by simulating datasets with changing sample sizes to show how REMI outperformed existing methodologies.
To contrast REMI with existing interactomes, the rebuilding of the lung adenocarcinoma (LUAD) interactome was zeroed in on. Different interpretations of the LUAD microenvironment have been assembled by utilizing different computational methodologies that have prompted novel insights:
- Interactome was assembled for LUAD utilizing mouse single-cell RNA sequencing data, where they utilized a scoring mechanism that captured highly expressed LR genes.
- Lung Tumor Microenvironment Interactome (LTMI) from bulk flow sorted RNA-seq information by thresholding gene expression levels in the dataset and calculating pairwise correlations between LR genes.
The REMI was applied to the LTMI dataset to recreate an interpretation of the LUAD interactome (REMI-LUAD) with higher particularity. Then, at that point, there was projected an inswinging LUAD scRNA-seq dataset onto REMI-LUAD to increase the cell type resolution of the interactome and alluded to this interactome as the single-cell rendition of REMI-LUAD (scREMI-LUAD).
Utilizing scREMI-LUAD, the paracrine interactions between cell subtypes were recognized, which were previously annotated as autocrine signaling interactions. To derive a signature of LUAD progression from scREMI-LUAD, prognostic score to every cell subtype and recognized prognostically related crosstalk signatures were assigned that might prompt clinically applicable biomarkers and remedial therapeutic targets.
REMI offers a newer approach to gathering cell-cell interactions between numerous cell types by representing conditional dependencies in cell-cell interactions. REMI is carried out in R and is uninhibitedly accessible on GitHub (https://github.com/plevritis-lab/REMI).
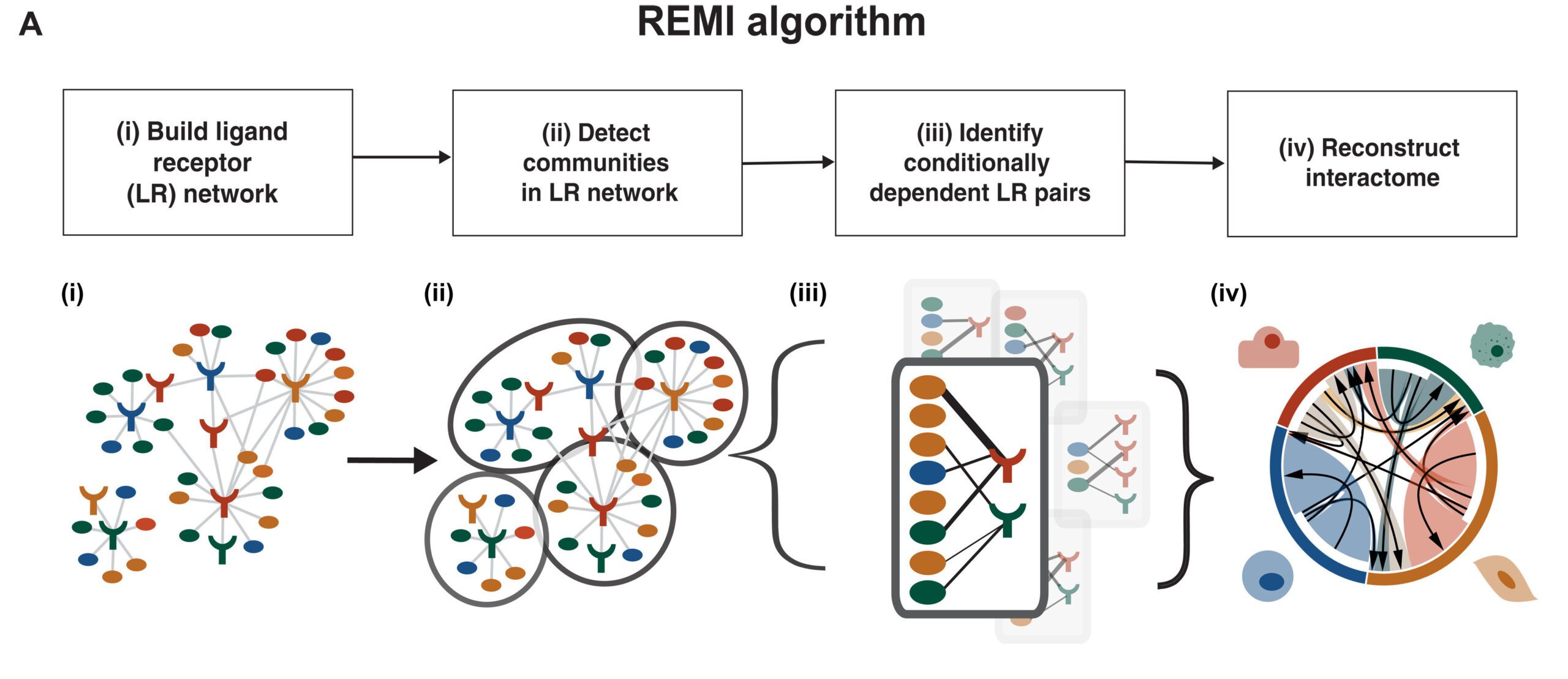
The REMI Algorithm
A modernized algorithm, REMI distinguishes communities of conditionally dependent cell-cell interactions to lessen the number of false-positive edges in LR correlation-based networks. The algorithm is appropriate for high-dimensional transcriptomic datasets with small sample sizes.
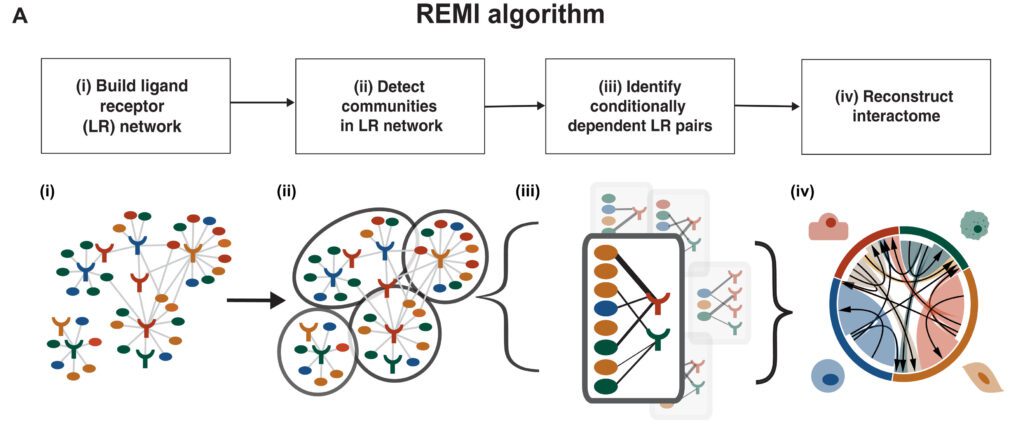
Image Source: Reconstructing codependent cellular cross-talk in lung adenocarcinoma using REMI
REMI is made out of four stages:
- Construct a weighted undirected LR correlation network by utilizing known LR pairings,
- Identification of communities of LR groups,
- Identification of conditionally dependent LR pairs in communities, and
- Rebuild the worldwide interactome from the communities
An extra advance in REMI considers the client to quantify the significance of an LR pair prediction concerning the LR pair’s REMI community.
In its initial step, REMI creates a weighted bipartite LR network, signified by G, where the nodes address either a ligand or a receptor gene expressed in a specific cell type inside the dataset. Edges of G are drawn between literature-supported LR pairings organized from the FANTOM5 database. Edge weights of G are calculated as the Pearson correlation between the gene expression of the LR nodes.
In ideal conditions, the conditional dependency of each LR pair would be calculated as for any remaining nodes in the network. In any case, current high-dimensional transcriptomic datasets contain countless numbers of genes contrasted with the number of samples, making the conditional dependency difficult to assess with high accuracy.
Robustness Testing for REMI Parameters via Simulations
To survey REMI’s presentation concerning executing GLasso alone, a populace level regularized interactome by utilizing the huge freely accessible Cancer Genome Atlas [The Disease Genome Atlas (TCGA)] LUAD bulk RNA-seq dataset was created. An assumption was made that the 1013 patients in this dataset address a whole populace and regularized a network of LR pairs inside the dataset utilizing GLasso. Considering the huge sample size, confounding impacts for all potential corporations inside the microenvironment were captured, and the subsequent organization contains these conditionally dependent LR pairs.
Increasing the Cell-Type Resolution of the REMI-LUAD by the Utilization of a scRNA-seq LUAD Dataset
To expand the cell type resolution of the REMI-LUAD, it was analyzed utilizing an openly accessible autonomous scRNA-seq LUAD dataset.
The cells were reclustered independently using the Louvain clustering method and applying the original copy’s annotated cell type labels. The resistant bunches with expansive resistant cell subtypes were marked from the original copy (myeloid, T cells, and B cells). For the leftover cell types, they distinguished five malignant, three endothelial, and four fibroblast subtypes.
To adjust REMI for scRNA-seq information, researchers arrived at the midpoint of every gene’s expression across every cell subtype per patient. The Lambrecht et al. dataset comprises just two patients with LUAD, which doesn’t give sufficient capacity to REMI’s inverse covariance-based calculations. Hence, they projected the scRNA-seq cell types onto GREMI−LUAD and relabeled the nodes’ cell types based on the expression levels in the single-cell dataset. This network alludes to single-cell REMI-LUAD (scREMI-LUAD).
For each subpopulation, the differentially expressed (DE) LR genes that had found the middle value of expression level more prominent than 0.4. Around 65% of the interactions in REMI-LUAD were available in scREMI-LUAD, affirming the high expression of these LRs in two autonomous patients. Since numerous LR genes show up in various cell subtypes, the quantity of interactions in scREMI-LUAD is more extraordinary than in REMI-LUAD, giving possible understanding into the degree of heterogeneity of the interactome.
For instance, REMI-LUAD inferred fibroblasts discharge CYR61, which interfaces with ITGB5 on fibroblasts, reminiscent of autocrine signaling. In scREMI-LUAD, CYR61 is expressed in F(2) and ITGB5 in F(4). Consequently, the REMI-LUAD CYR61:ITGB5 autocrine fibroblast interaction is re-defined as paracrine signaling between various fibroblast cell subtypes in scREMI-LUAD.
Note that 86% of autocrine signaling interactions were changed over into paracrine interactions between cell subtypes. This expanded number of paracrine interactions features the degree of the perplexing crosstalk in the TME.
The Endpoint
REMI offers the real benefit of distinguishing co-dependent interactions on a global scale with a small sample size by utilizing previous knowledge, network analysis, and sparsity principles. REMI’s performance on an enormous transcriptomic dataset from TCGA shows that estimating the inverse covariance matrix using REMI is robust.
REMI’s performance was estimated across changing sample sizes, and observed that REMI’s specificity decreased less than its sensitivity relative to GLasso. REMI likewise beats existing cell-cell interactions, and derivation draws near. The distinction in performance measurements can be made sense by the assumptions fundamental to every calculation. CellphoneDB v2.0 predicts LR pairs based on the mean expression values, as higher LR genes are more likely to be expressed and interaction-efficient. NicheNet depends on literature-derived models and user-defined inputs to indicate downstream activated genes.
REMI centers around capturing the straight transcriptional connection between a ligand and a receptor, which isn’t also captured in other techniques. Generally speaking, REMI’s higher specificity is positive while choosing interactions for experimental validation to decrease the risk of false positives.
Story Source: Yu, A., Li, Y., Li, I., Ozawa, M. G., Yeh, C., Chiou, A. E., … Plevritis, S. K. (2022). Reconstructing codependent cellular cross-talk in lung adenocarcinoma using REMI. Science Advances, 8(11), eabi4757. https://doi.org/10.1126/sciadv.abi4757
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.





{kind=link}