
The iconic bacterial genus from sheep rumen ‘Quinella’ has been the center of bacterial taxonomy for various reasons. The research group from Massey University, New Zealand, and the National University of Singapore have deciphered the genomic insights using metagenomic sequencing. The analysis provided observations into taxonomic diversity and assess genetic data for the physiological understanding of the unculturable gut microbe.
Introduction
In a recent decade of microbiology and biomedical research, understanding and deciphering the gut microbiome has emerged due to its potential and contribution towards healthcare and welfare. Considering the prevalence of gut microbes in the rumen of cattle and sheep has been of greater importance since the health benefits of digestion and development are no different from humans. Isolation and culturing, the classical identification is primarily considered to obtain these microbes in pure cultures for better insight.
One of the iconic anaerobic unculturable bacteria from sheep rumen ‘Quinella’ was first reported in 1913 and still remains an unculturable entity. Some previous research states its possible connection with low methane emission among Quinella-rich sheep rumen. The ‘Quinella’ is closely related to the members of the Selenomonas-Megasphaera- Sporomusa group of bacteria, classified in the order Selenomonadales in the class Negativicutes. While the possible network between the two has not been established, the group of researchers from Massey University, New Zealand, and the National University of Singapore has elucidated the metagenomic data from publicly available databases and animal sampling under ethics approval.
Image source: https://doi.org/10.1038/s41467-022-34013-1
Considering the overall population dynamics of ‘Quinella’ detected in the rumen ranged around 2.3 to 4.5 % of bacteria assessed using 16srRNA gene abundance. While the low methane-yielding communities, the Q-type was evidently rare in cattle populations but accounted for about 32.5 %. This influenced understanding the earlier relatives and deciphering the physiological characteristics based on genetic understanding.
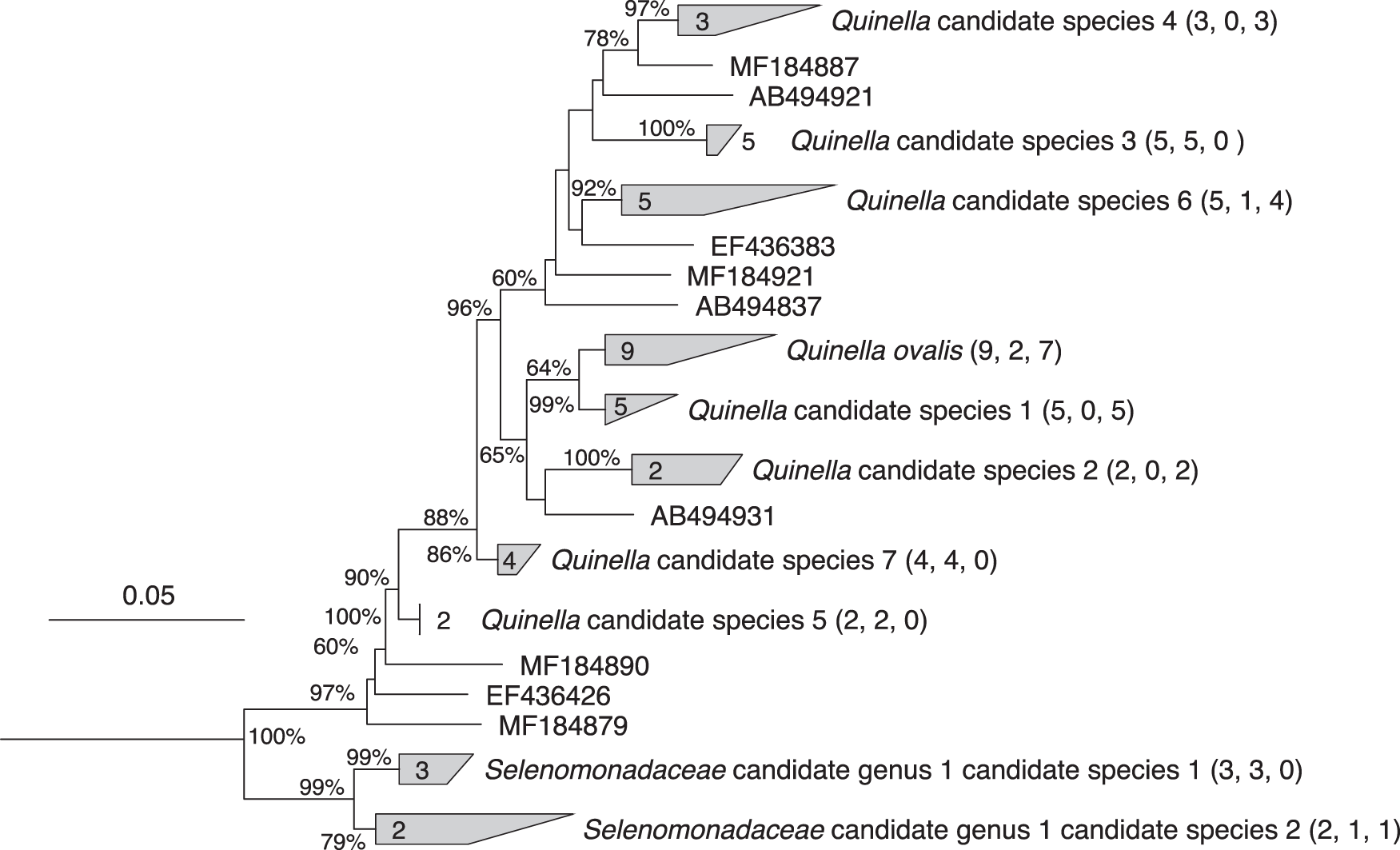
The methodology was based on understanding the taxonomic definition of unculturable anaerobe, providing an understanding of the diversity of Quinella species, using high-intensity microscopy to understand bacteria’s ultrastructure, constructing the genomic bins and deducing the fermentation pathway of Quinella spp. Researchers, in order to obtain the full length of 16srRNA gene sequences from Quinella spp. With a 97% similarity index for species for 93% similarity for genera, across the database, Six of the newly cloned Quinella sequences clustered with the reference sequence of Q.ovalis (GenBank accession M62701). The analysis suggested the presence of multiple species of the genus. While a potential new genus level cluster (Selenomonadaceae candidate genus 1) with the relativity of >92.4% was also seen.
The team adeptly put efforts into the physical enrichment and comparison of Quinella cells from rumen samples. These samples were processed using a modification of protocol physical enrichment from Wicken and Howard’s method. The diversity of the anaerobe species from enriched samples was investigated using a total of 155 quinella-16srRNA cloned genomic sequences from the three samples, i.e., pooled samples from 12 sheep, two weeks apart pooled samples from one sheep, and single rumen sample from one sheep which allowed reconstruction of genomes from extracted DNA to elucidate on metabolic potential.
In order to facilitate the concentration of Quinella species within the rumen, the team suggested FISH (fluorescence in situ hybridization) using DNA probes specifically designed to match 16srRNA – Quin1231 and EUB338, which binds to 16S rRNA from most of the known bacterial species. Probing and phase contrast imaging resulted in the distinguished identification of oval cells as supposed to be fluorophore associated.
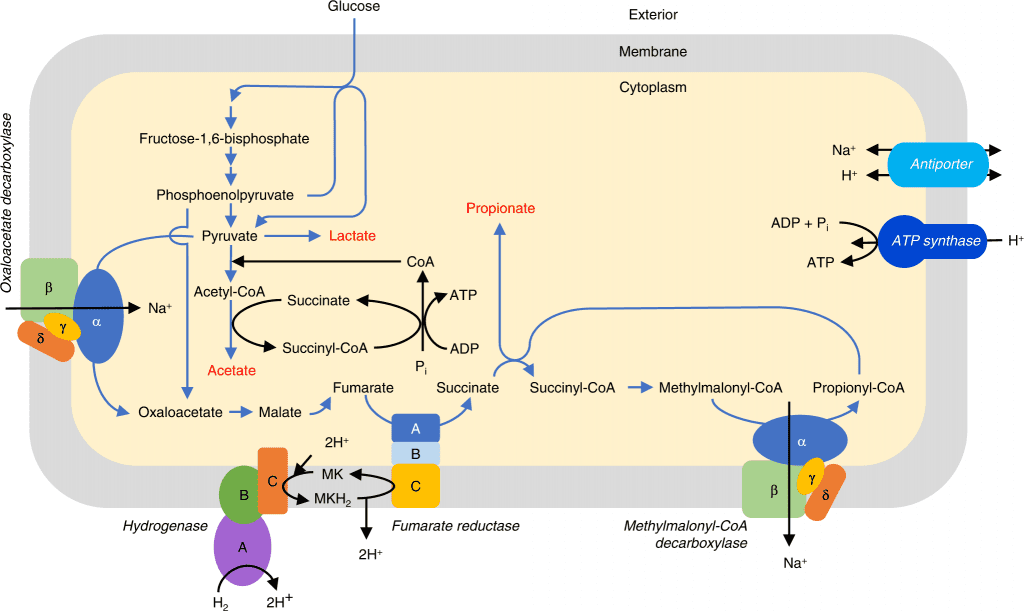
Image Source: https://doi.org/10.1038/s41467-022-34013-1
The three samples of ‘Quinella cells’ were used to construct the genomic bins, generated metagenome-assembled genomic DNA extracted from samples, and sequenced using Illumina MiSeq Technology. A total of thirteen bins were identified, and four bins were considered nearly complete >90% with medium to low contaminations. The total bins selected were bin 5 from the sample 1 (designated bin SR1Q5), bin 7 from the sample 1 (SR1Q7), bin 5 from the sample 2 (SR2Q5), and bin 1 from the sample 3 (SR3Q1).
In order to understand the metabolic activity, an analysis of four genome bins for the Quinella species was constructed, and CAZymes analysis was primarily suggested. Using the understanding of gene coding, the Quinella genus contained genes for L-lactate dehydrogenase, suggesting the end product as lactase, pyruvate: ferredoxin oxidoreductase, which converts the pyruvate to acetyl-CoA. At the same time, it does not appear to produce formate and ethanol as the end products. The analysis has indicated the use of randomized pathways for propionate formation in Quinella.
Final Thoughts
The overall understanding of these pathways, such as hexose fermentation to propionate, appears very similar to those of other facultative anaerobes such as Prevotella. The construction of genome bins has elucidated in depth the understanding of metabolic pathways and overall physiological traits using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). Further understanding of genetic structures and the construction of genomic bins will help determine the environmental landscapes for the rumen-microbiome and provide details on culturing the bacteria in their purest form.
Article Source: Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Mahi Sharma is a consulting Content Writing Intern at the Centre of Bioinformatics Research and Technology (CBIRT). She is a postgraduate with a Master's in Immunology degree from Amity University, Noida. She has interned at CSIR-Institute of Microbial Technology, working on human and environmental microbiomes. She actively promotes research on microcosmos and science communication through her personal blog.





{kind=link}