
A low-cost metagenome sequencing technology has been developed by scientists from the Chinese Academy of Sciences’ Qingdao Institute of Bioenergy and Bioprocess Technology (QIBEBT). It allows high-resolution disclosure of a microbiome’s whole ‘landscape’ of species, even from low-biomass, damaged, or polluted samples.
The ‘microbiome‘ is referred to a community of microorganisms (bacteria, fungi, protists, and viruses) that live in and on people, as well as any other environment. To determine which microorganisms are part of a specific microbiome, researchers must do metagenomic sequencing, the simultaneous sequencing of genetic material from numerous species.
Even state-of-the-art metagenomic sequencing, however, has a lot of critical drawbacks. The procedure is divided into two categories: amplicon sequencing and whole metagenome sequencing (WMS).
Amplicon sequencing is a low-cost procedure. Unfortunately, it only provides information on microbiome members at the genus level of the taxonomy. It is difficult to achieve taxonomic information at the species or strain level. Amplicon sequencing also has the drawback of detecting bacteria, archaea, fungus, and viruses individually rather than identifying them all at once.
WMS can capture whole DNA sequences of all organisms in a sample, including bacteria, archaea, fungi, and viruses, and at a higher taxonomic resolution. This method provides a lot more information, but it’s also a lot more expensive. Moreover, WMS also demands a significant volume of high-quality DNA.
“What we need is a low-cost sequencing method that can enable accurate, species-resolution identification of all microbes at the same time and can handle low biomass samples, ” revealed SUN Zheng, first author of the paper and a researcher with the Single-Cell Center at QIBEBT.
The researchers discovered the “2bRAD sequencing for Microbiome” technology, often known as 2bRAD-M. This makes use of restriction site-associated DNA sequencing (RADseq), which digests genomic DNA from feces, skin, or environment-surface materials into fragments of 25-33 base pairs (pairs of nucleotides) using Type IIB restriction enzymes (proteins that cleave DNA at specific locations along the molecule). Only these digested fragments—roughly 1% of the metagenome—are sequenced using the 2bRAD-M approach, which can yield profiles spanning bacteria, archaea, and fungus, and not only up to genus but up to species level.
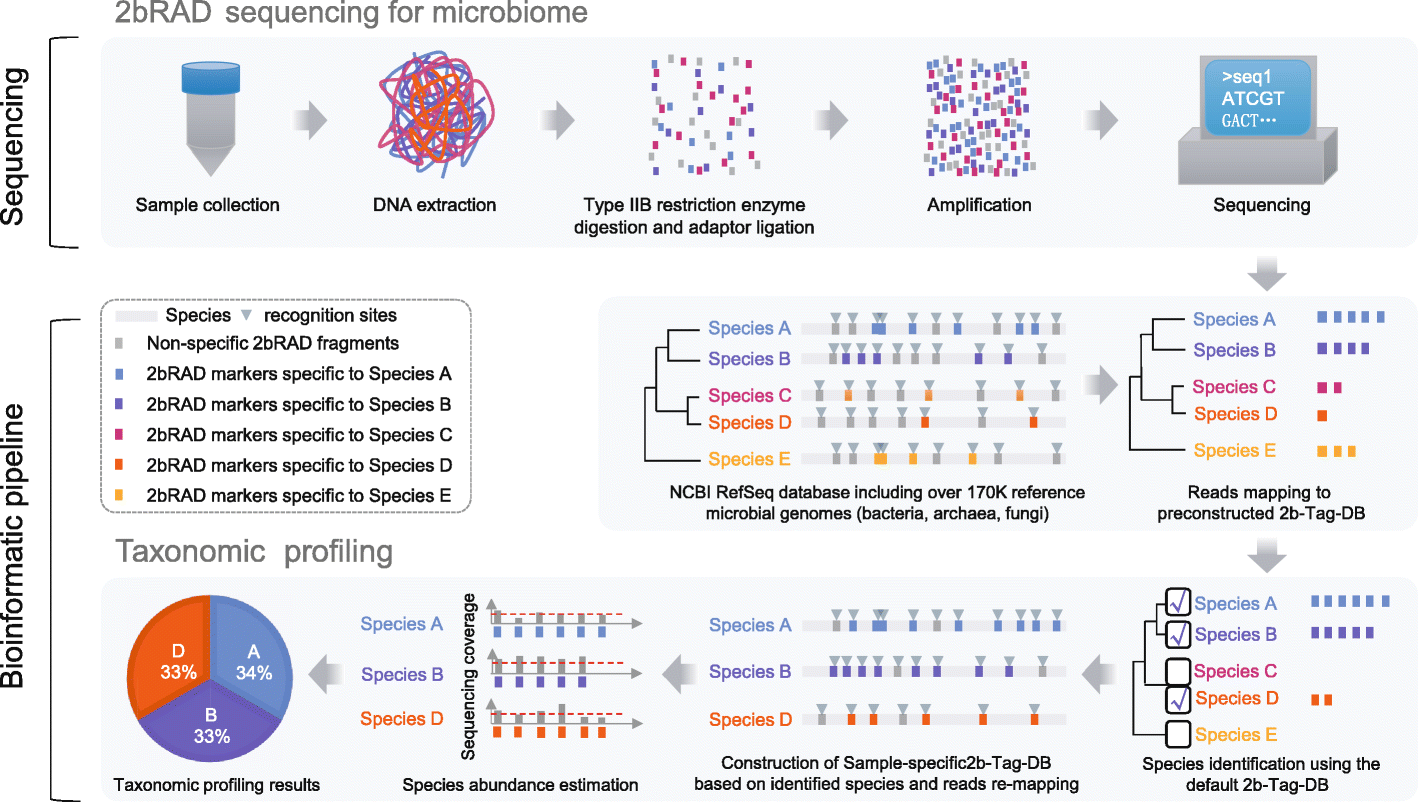
“By needing only a single picogram (a thousandth of a nanogram) of total DNA, the process can cheaply and accurately generate high-resolution profiles even for hard-to-sequence samples where there has been high host DNA contamination, or the DNA is severely fragmented or otherwise degraded,” stated HUANG Shi, co-first author of the paper.
The involvement of microbes in cancer formation has been one of the most exciting discoveries in recent years. Using 2bRAD-M, the researchers demonstrated that the microbiome profiles from cervical cancer tissues might be revealed from formalin fixed paraffin embedded (FFPE) samples, which are often poor in quantity, damaged, or contaminated by human DNA. Such profiles might be used to aid in the early detection of cervical malignancies.
“In the future, we hope to further develop the 2bRAD-M technique and explore its various clinical applications,” told senior author XU Jian, Director of Single-Cell Center at QIBEBT.
“Ultimately, we hope that 2bRAD-M would become a cost-efficient, popular tool for microbiome research and conquer various challenging tasks that are beyond the reach of current techniques,” concluded Prof. WANG Shi from the Ocean University of China, another senior author of the study.
Story Source: Sun, Z., Huang, S., Zhu, P. et al. Species-resolved sequencing of low-biomass or degraded microbiomes using 2bRAD-M. Genome Biol 23, 36 (2022). https://doi.org/10.1186/s13059-021-02576-9
http://english.qibebt.cas.cn/ne/rp/202201/t20220125_299843.html
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











{kind=link}