
Scientists from the University of Minnesota, Minneapolis, USA, with their collaborators, developed a machine learning framework for a multi-omic integration to identify the shared and disease-specific host gene microbiome associations across human diseases. They identified the associations between gut microorganisms and host genes that portray shared as well as disease explicit patterns.
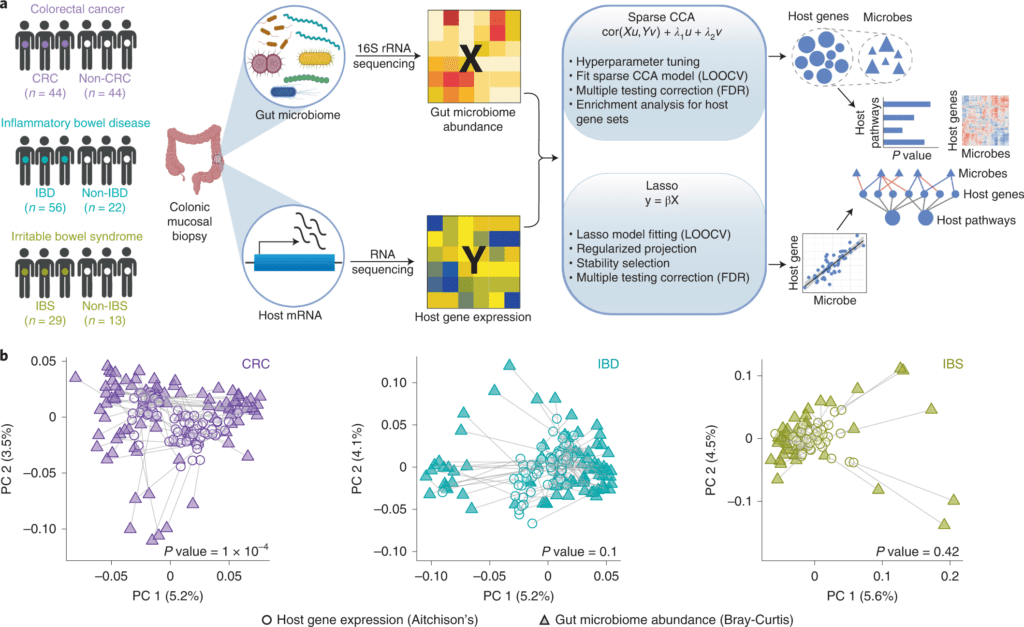
Image Source: Identification of shared and disease-specific host gene–microbiome associations across human diseases using multi-omic integration.
While gut microbiome and host gene regulation contribute to gastrointestinal disorders autonomously, there is still little known about how the two might interact to affect host pathophysiology.
For this purpose, the scientists developed a machine learning-based framework for the joint analysis of paired host transcriptomic (n = 208) and gut microbiome (n = 208) profiles from colonic mucosal samples collected from the patients with colorectal cancer, inflammatory bowel disease, and irritable bowel syndrome.
They identified the associations between gut microorganisms and host genes that portray shared and disease explicit patterns and found that a typical set of host genes and pathways involved in gastrointestinal inflammation, gut barrier protection, and energy metabolism are related to disease explicit gut microorganisms.
Furthermore, they likewise found that mucosal gut microbes that have been implicated in every one of the three illnesses, like Streptococcus, are related to various host pathways in every disease, proposing that similar microorganisms can influence host pathophysiology in a disease explicit way through regulation of different host genes.
The framework developed can be applied to different diseases for the identification of host gene microbiome associations that might impact disease results.
Requirement for Clarity in the Determination of How the Factors Affect Host Pathophysiology
The human gut microbiome assumes a basic role in the modulation of human health and disease. Alterations in the composition of the human gut microbiome have been related to a wide range of chronic diseases, such as colorectal cancer (CRC), inflammatory bowel disease (IBD), and irritable bowel syndrome (IBS).
For instance, past studies have reported an expansion in the abundance of Fusobacterium nucleatum and Parvimonas in CRC, decreased abundance of Faecalibacterium prausnitzii, and enrichment of enterotoxigenic Bacteroides fragilis in CRC and IBD, and overrepresentation of Enterobacteriaceae and Streptococcus in IBD and IBS.
Notwithstanding the gut microbiome, dysregulation and pathways have additionally been implicated in these diseases.
Scientists have reported the disruption of Notch and WNT signaling pathways in CRC and activation of toll-like receptors (for instance, TLR4) that incite NF-κB and TNF-α signaling pathways in IBD dysregulation of immune response and intestinal antibacterial gene expression in IBS.
While host transcription and gut microbiome have independently been distinguished as contributing factors to gastrointestinal (GI) diseases, it is unclear the way that the two might associate to impact host pathophysiology.
Unraveling the Role of Gut Microbiome and Host Gene Regulation in Pathogenesis
Studies in model life forms have indicated that the regulation of gene expression by the gut microbiome in the host is a potential mechanism by which organisms can influence host physiology.
For instance, in zebrafish, the gut microbiome adversely regulates the transcription factor hepatocyte nuclear factor 4, prompting host gene expression profiles related to human IBD.
The gut microbiota can modify host epigenetic programming to modulate intestinal gene expression engaged with immune and metabolic processes in mice.
Also, the latest in vitro cell culture experiments have demonstrated the way that particular gut microbes can alter the gene expression in interacting human colonic epithelial cells.
Given the evidence for crosstalk between the gut microbiome and host gene regulation, characterizing the interplay between the two variables is fundamental for explaining their part in the pathogenesis of human intestinal diseases.
Studies to Reveal Insights on Host Gene-Microbiome Crosstalk
A couple of latest studies have researched the associations between the host transcriptome and gut microbiome in specific human gut disorders, including IBD, CRC, and IBS.
For instance, studies analyzing microbiome-host gene connections in IBD have identified mucosal microbiome associations with host transcripts enriched for immunoinflammatory pathways.
Studies exploring the role of host gene microbiome association in CRC have found connections between the abundance of pathogenic mucosal bacteria and the expression of host genes involved in gastrointestinal irritation and tumorigenesis.
In IBS, host genes involved in gut barrier function and peptidoglycan binding, like KIFC3 and PGLYRP1, are related to the microbial abundance of Peptostreptococcaceae and Intestinibacter.
While the examinations have uncovered significant insights about host gene-microbiome crosstalk in GI diseases, they are restricted in a few viewpoints.
For instance, most studies have inspected the association between a restricted subset of host genes and gut microorganisms; for example, by focusing just on differentially expressed genes, those related to immune functions, or select microorganisms representing bacterial clusters or co-abundance groups, consequently characterizing just a subset of potential associations.
What’s more, the identification of host gene-microbe association depends on testing for pairwise correlation between the host’s gene and microorganism by the utilization of Spearman or Pearson correlation, subsequently overlooking the innate multivariate properties of the datasets.
Moreover, most studies center around inspecting associations in a solitary disease at once, and remarkable examples of host-microbiome association across numerous disease states remain ineffectively characterized.
Characterization of Disease-Specific and Shared Host-Gene Microbiome Associations
In this study, the scientists comprehensively demonstrated the association between the mucosal gene expression and microbiome composition in patients with colorectal cancer, inflammatory bowel disorder, and irritable bowel syndrome — three GI problems in which both host gene regulation and gut microbiome have been implicated as contributing factors.
They developed and applied a machine learning framework that overcomes average difficulties in multi-omic integrations, including high-dimensionality, sparsity, and multicollinearity, to distinguish biologically significant associations between gut microbes and host genes and pathways in every disease.
They utilized the system to demonstrate disease explicit and shared host gene-microbiome association across the three diseases that might facilitate insights into the molecular mechanisms underlying the pathophysiology of gastrointestinal diseases.
The Endpoint
This work shows the power of incorporating gut microbiome and host gene expression information to give bits of knowledge into their combined role in GI diseases, including CRC, IBD, and IBS.
Taken together, the outcomes indicate that GI diseases are characterized by a complex network of associations among microorganisms and host genes.
Albeit these associations can be disease explicit, there are cases where a similar microbial taxon is related to different host genes in every disease, as well as the other way around: cases where a similar host pathway is connected to various microorganisms in every disease.
Albeit much effort in microbiome research has been coordinated towards identifying explicit microbial taxa that are liable for the pathogenesis of illness, the scientists’ findings demonstrate that it is essential to integrate host genomics information, as it can give important data on the potential mechanisms through which microorganisms can influence health.
The outcomes represent a significant stage toward characterizing the association between the gut microbiome and host gene regulation and understanding the contribution of the microbiome to disease etiology.
Article Source: Priya, S., Burns, M.B., Ward, T. et al. Identification of shared and disease-specific host gene–microbiome associations across human diseases using multi-omic integration. Nat Microbiol (2022). https://doi.org/10.1038/s41564-022-01121-z
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}