
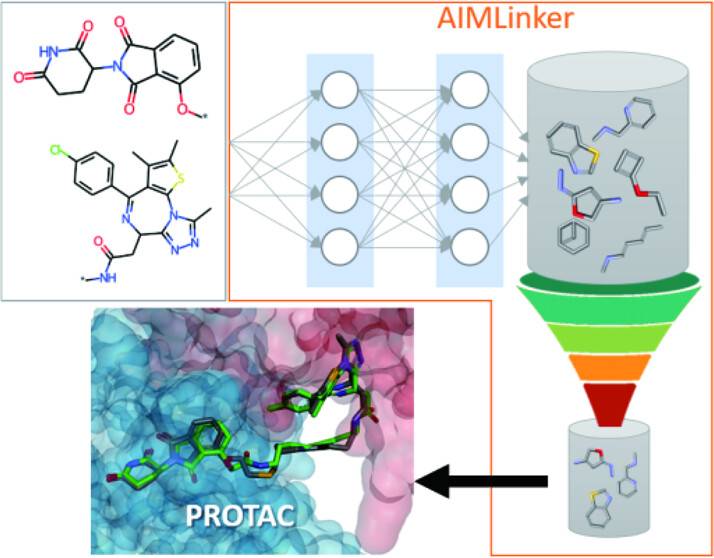
PROTACs (PROteolysis TArgeting Chimeras) are small molecules capable of degrading target proteins. They have wide applications in designing drugs with therapeutic effects, especially in the case of cancer. But, conventional drug design is a complex and tedious process involving multiple iterations to obtain optimized results. Hence, scientists from AnHorn Medicines Co., Ltd., Taiwan, have proposed the application of AIMLinker, a deep-learning-based neural network, to simplify and accelerate the generation of meaningful PROTAC analogs. The AIMLinker model collects structural information from the input fragments and generates linkers (chemical components to connect different parts of a compound) to join the input fragments and yield a cohesive structure. The generated PROTAC analogs show better binding affinity in comparison to the already existing PROTAC molecules created from conventional drug design processes.
Drug Design and PROTACs: A Match NOT Made in Heaven?
Conventional drug design processes typically involve structure-based drug design. It not only consumes a large search space but, to yield the most optimized results, multiple iterations concerning binding affinities, molecular structures, and pharmacokinetics have to be conducted. Yet, it is an essential process because drug discovery has its limitations, and structure-based drug design can lead to the discovery of a drug lead. A drug lead is not a drug in itself but a compound with a higher nanomolar affinity toward a target. Thus, the discovery of a drug lead is vital to facilitating efficient drug discovery.
Recently, PROTACs, small heterobifunctional molecules capable of inducing the degradation of target proteins, have gained popularity. They consist of two integral components: a ligand for securing the target protein of interest (POI) and a ligand for ubiquitin-protein ligase (E3, involved in the degradation of other proteins within the cells). Linkers link these two components together. Degradation occurs when POI and E3 form a ternary complex.
From a structure-based drug design point of view, designing PROTACs involves identifying the most suitable combination of the three chemical entities (POI, E3, and Linker). Using conventional methods, Winter et al. have designed dBET6, which causes the proteasomal degradation of BRD4. BRD4 is a protein belonging to BET (Bromodomain and Extra Terminal) family and is involved in causing cancer by regulating oncogenes’ expression and organizing super-enhancers. The E3 ligand here is CUL4-RBX1-DDB1-CRBN E3 ubiquitin ligase, and the course of action for the degradation of BRD4 is its linkage with BET proteins.
Yet, with the structure-based drug design of PROTACs, the design and generation of linkers still need to be clarified and require further investigation.
Is Deep Learning the Key to Unlocking Better PROTACs Design?
Owing to the shortcomings of structure-based drug design, there arises the need for a tool or technology that can speed up the generation of PROTAC analogs, design better linkers as well as improve the chemical properties of PROTACs.
But before that, let’s discuss the benefits of PROTACs. In comparison to conventional occupancy-driven small molecule inhibitors, PROTACs have several advantages, including a catalytic nature, reduced dosage and dosing frequency, a stronger and long-lasting effect, an additional layer of selectivity to lower potential toxic effects, the ability to overcome drug resistance, the ability to target nonenzymatic functions, and extensive target space.
Recent studies throw light on the efficacy of deep-learning algorithms in the discovery of novel molecular structures. The results are pretty promising as far as accurate screening of potential drug targets is concerned. Graph Neural Networks (GNN) are emerging as a vital drug discovery method. It utilizes graph convolutions to learn task-specific representations while preserving the atom-bond interactions automatically. In contrast to descriptor-based models, GNN models show much better performance predicting properties. Gated Graph Neural Network (GGNN), a variant of GNN, effectively encodes molecular structures and allows the formation of new structures with desired characteristics. Even fragment linker designing techniques such as DeLinker and 3DLinker backed by deep neural networks have emerged for molecular linkers.
However, these existing approaches focus more on two-dimensional representation and fail to consider three-dimensional information, which may negatively impact the design process. Also, they lack methods capable of refining the generated molecules and validating them based on molecular conformations.
AIMLinker: Can it Emerge as an Antidote to the Existing Difficulties in PROTACs Design?
The researchers have proposed Artificial Intelligence Molecule Linker(AIMLinker) to solve the existing problems. AIMLinker is a deep-learning-based neural network that integrates the designing, generating, and screening of small molecular structures for PROTACs linker into a unified framework, thus increasing efficiency and effectiveness. AIMLinker considers the 3D structural information of two input fragments. The information contains details about angles, distances, and spatial positions. AIMLinker employs the GGNN method as its core architecture, representing atoms as nodes and bonds as edges for modeling the molecular structure. It constructs the model in a step-by-step manner through an iterative process in which atoms are added, and bonds are formed until termination. Then, the generation of the molecular structure is followed by a readout step and then a validation step. The validation is done using techniques such as docking, root-mean-square deviation (RMSD), relative Gibbs free energy, molecular dynamics (MD) simulation, and free energy perturbation (FEP) simulation. This helps in assessing the suitability of the molecules as potential drug candidates. AIMLinker presents an end-to-end pipeline that covers all the above processes in a single platform.
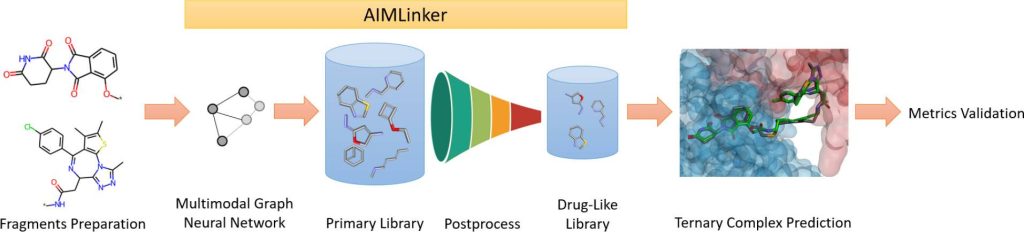
Image Source: https://doi.org/10.1021/acs.jcim.2c01287
Detailed Process of PROTACs Design Using AIMLinker
The first step is pre-processing, which involves selecting and preparing protein and ligase structures to be fed as the inputs in the encoder-decoder network. A multimodal encoder-decoder network, trained with a variational autoencoder (VAE), generates structural linkers between input fragments. The training dataset contains molecules from the ZINC and PROTAC-DB databases, segregated into fragments and linkers. Hyperparameters are optimized to improve the model’s performance. Post-processing steps are implemented, which filter and refine the model outputs, and remove duplicates, unfavorable substructures, and molecules violating Bredt’s rule. Then AutoDock4 software predicts the best binding poses of PROTACs by considering binding energy, biochemical properties, and entropy. Metrics such as RMSD and binding affinity are calculated for both the generated molecule and the reference molecule. MD and free energy perturbation simulations are conducted to evaluate the chemical properties of the generated molecule relative to the reference compound. The reference compound used is dBET6.
Comparative Analysis and Superiority of Generated Molecules Over dBET6
The most optimized out of all the generated molecules are compared with dBET6, and the following observations can be made:-
- The generated molecules exhibit ring-like structures as opposed to dBET6’s linear structure. Hence, the generated molecules have more stability and a better ability to form π bonds in binding pockets.
- Docking with AutoDock4 showed that the generated molecules have better binding affinities than dBET6.
- The free energy perturbation simulations confirmed the superior binding affinity of the most stable generated molecule (6BOY_1268) compared to dBET6.
Conclusion
Drug discovery and design are crucial components of bioinformatics for the welfare of humankind. The easier, faster, and more optimized it becomes, the better. This study proposes the use of AIMLinker, a deep-learning-based platform, to generate better PROTAC analogs in a faster and more efficient manner. AIMLinker provides an end-to-end pipeline for conducting pre-processing, design, and screening of molecules in a single platform. The generated molecules have better chemical properties than the existing ones. Even though the current generation has its shortcomings, as the study focuses only on a single PROTACs target for testing and validating the proposed model, there is scope for improvement and an aim for expansion of the utility of the model by applying it to more PROTACs targets and investigating its applications in other structure-based drug discovery and design approaches.
Article Source: Reference Paper | AIMLinker: Code Availability
Learn More:




Neegar is a consulting scientific content writing intern at CBIRT. She's a final-year student pursuing a B.Tech in Biotechnology at Odisha University of Technology and Research. Neegar's enthusiasm is sparked by the dynamic and interdisciplinary aspects of bioinformatics. She possesses a remarkable ability to elucidate intricate concepts using accessible language. Consequently, she aspires to amalgamate her proficiency in bioinformatics with her passion for writing, aiming to convey pioneering breakthroughs and innovations in the field of bioinformatics in a comprehensible manner to a wide audience.





{kind=link}