
Protein identification has gained much importance over the last few years. In the shotgun proteomics-based approach, peptide fragments are used to identify proteins. The quantification of proteins from identified peptides is a difficult task. Scientists have suggested ProInfer, a new machine learning-based approach for performing Protein Inference based on biological networks.
Mass Spectrometry (MS)-based Proteomics
Mass spectrometry (MS)-based proteomics study is used to analyze the protein expression in an organism. Lately, a shotgun proteomics-based approach has been used to quantify proteins. It identifies and quantifies proteins in a sample using tandem mass spectrometry and liquid chromatography methods. First, the protein is digested into a mixture of peptide sequences. The peptides are subsequently isolated using liquid chromatography (LC). The peptide mixture is then scanned by tandem mass spectrometry (MS/MS) to obtain a collection of MS/MS spectra.
For peptide identification, fragments undergo ionization to generate Peptide-Spectrum Matches (PSM), which are analyzed via peptide identification algorithms. The peptides thus identified are assembled for identification, and the process is termed Protein Inference. Tools like Percolator and PeptideProphet can perform peptide identification.
The Protein Inference and Challenges Faced
Protein Inference refers to the process of protein identification by identifying peptides in a biological sample. Protein Inference is a tedious task, and slight deviations in experimental or computational methods can result in false values. Several tools have been developed to perform Protein Inference, like Percolator, EPIFANY, FIDO, PIA, ProteinProphet, etc.
Computational or analytical problems while performing Protein Inference may occur due to the design of the algorithm, the use of different parameters, or sequence library-related issues. Experimental problems include incomplete digestion of proteins, problems in protein separation and ionization, and the sensitivity and resolution of the spectrometer, to name a few. Apart from this, the presence of degenerate peptides and one-hit-wonder proteins also causes problems in protein identification. One-hit wonder proteins have only a single identified peptide, and degenerate peptides have peptide fragments that can be shared by several proteins, which makes protein identification difficult.
Uncertainty due to peptide sequences can be resolved either by removing the dubious peptide using tools like Percolator or performing network-wide peptide-protein analysis using tools like EPIFANY. A semi-supervised machine learning method was used to train Percolator, which analyzes the spectrum generated by the post-processing of peptide-spectrum matching. Percolator avoids the use of deviant peptides to increase its efficiency.
To resolve the problem arising from deviant peptides, scientists have developed a tool called Fido, based on Bayesian probability. Similarly, PIA is another tool that integrates results from various different protein inference tools. It tackles the problem of deviant peptides by using the maximum parsimony principle to generate minimum sets of peptides. EPIFANY uses a convolution tree-based machine learning algorithm for protein identification. These tools are efficient in protein identification, but efficient and accurate proteome coverage for a biological sample still needs improvements.
Since these tools are trained to identify direct peptides to protein confirmation, they often ignore proteins with low peptide content in small quantities. Scientists have proposed a new protein inference tool called ProInfer (short for Protein Inference), which uses a simple protein scoring algorithm based on probability accumulation. Its algorithm is based on a previously existing algorithm, PROTREC. PROTREC relies on the concept that proteins that form stable complexes with other proteins are likely to be expressed together.
ProInfer (Protein Inference)
ProInfer efficiently performs protein inference as well as effectively analyzes the protein quantitatively. ProInfer is based on the peptide-protein network, which is the representation of peptide-protein interactions and proves the presence of our query protein in the test sample. The peptide-protein network helps us understand how protein complexes are formed from peptides.
Protein Inference models are trained to read data from spectral analysis. At times, constraints arising due to the spectrometer’s sensitivity and the lack of protein in the sample can cause weak signals, making them difficult to observe. To overcome this problem in proteins, the algorithm is trained to obtain information from peptide-protein networks. The PROTREC tool can extract information on proteins with low peptide support.
While designing ProInfer, scientists assumed that protein complexes were important in understanding and improving the quality of Protein Inference. The basic functioning of ProInfer can be summed up as the following three steps:
1. Peptide-protein network-based Protein Inference.
2. Inference of protein complex.
3. Protein Rescoring based on Protein-complex networks.
The ProInfer algorithm is based on calculating the Maximum Posterior Probability of constituent protein. ProInfer conducts peptide-spectrum match (PSM) on identified Peptide fragments, resulting in Protein Inference. The peptide-spectrum match is analyzed and processed by a Posterior Error Probability (PEP) score. A strong posterior error probability has higher certainty for peptide-spectrum match than a relaxed one.
Validation and Limitations
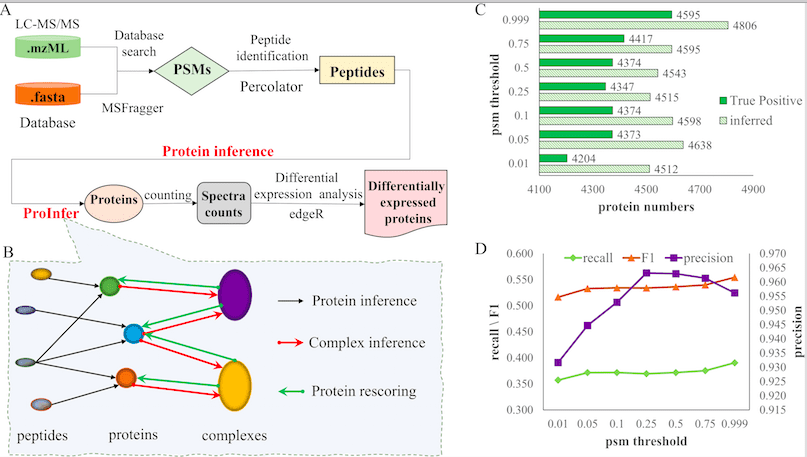
To validate the data generated, scientists used ProInfer along with other similar tools like EPIFANY, Percolator, and PIA was used to perform Protein Inference on Hela Cell Line. The result showed ProInfer calculated the greatest number of true positives inference. ProInfer was able to identify a higher number of proteins compared to the other tools. ProInfer could also identify differentially expressed proteins. These data validated the superiority of ProInfer compared to other tools of the same kind. It also highlighted ProInfer’s ability to access information from biological networks and protein complexes to process peptides.
ProInfer has some limitations and needs to be fine-tuned for better results.
1. ProInfer is dependent on the biological network to deduce its result. The number of organisms with a well-characterized protein-complex database is very low, thus limiting the functionality of ProInfer.
2. ProInfer still needs to be optimized to enhance its performance. Currently, only a subset of protein is considered for calculating the probability. Low coverage corresponds to lower performance.
3. ProInfer is trained using protein complex databases. In the case of paralogous proteins, which share the same peptide to form protein complexes ProInfer identifies them as absent, thus giving a false positive value.
Future Implications and Conclusions
The complete working principle and algorithms of ProInfer were published in the journal PLOS Computational Biology. Scientists have found ProInfer, reliable and efficient in identifying proteins compared to similar tools. They also suggested ProInfer may be utilized for profiling the protein function and may be used for identifying drug targets and biomarkers.
Article Source: Reference Paper
Learn More:




Sipra Das is a consulting scientific content writing intern at CBIRT who specializes in the field of Proteomics-related content writing. With a passion for scientific writing, she has accumulated 8 years of experience in this domain. She holds a Master's degree in Bioinformatics and has completed an internship at the esteemed NIMHANS in Bangalore. She brings a unique combination of scientific expertise and writing prowess to her work, delivering high-quality content that is both informative and engaging.





{kind=link}