
Scientists at the Institute of Parasitology and Biomedicine and the University of Granada, Spain, collaborated to develop two pipelines that could automate and optimize metagenomics and metatranscriptomics data analysis. These pipelines could be adapted for 16S, shotgun, and RNA-Seq data. Its performance was validated through three studies by assessing their taxonomy classification ability.
When Anton van Leeuwenhoek first opened the doors to the unseen world of microorganisms in 1673 through his self-made single-lens microscope, it couldn’t have been possible to imagine the explosion of discoveries that were to follow in its wake. The paradoxical world of microbes is a source of infinite curiosity to many scientists around the world. Thus, it was a no-brainer that with the advent of NGS, the microbes would get their very own niche within it—Metagenomics.
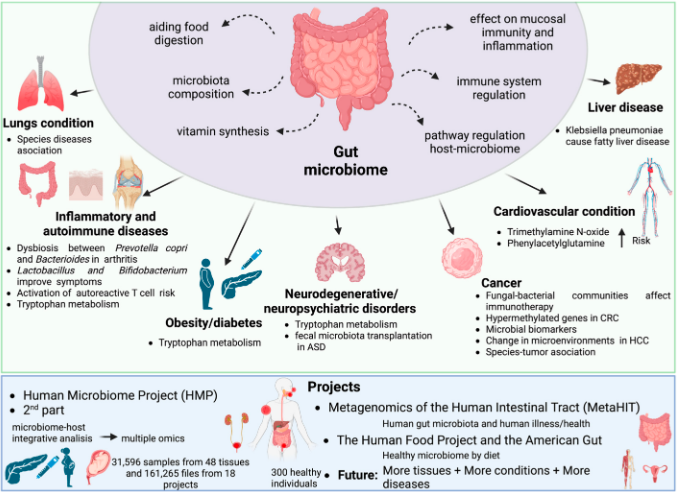
Metagenomics enables the analysis of the microbial population within a sample based on the genetic material isolated from it. The role and modulation of microbiomes in driving and assisting various changes within the environment they exist in, especially that of the human body, is heavily studied. Studies in the past few decades have revealed the influence of microbiota in many diseases and their treatment. Over 4,300 articles have been published in the past five years on gut microbiota, as reported by PubMed.
Metagenomics data analysis is a maze laid down by large quantities of data and an array of computerized tools. To navigate through it successfully, one has to choose the correct tools at each step based on the sequencing method, computational resources, and goal of the study, which is very tedious and complex. Pipelines that string together tasks and perform them automatically allow for alleviating these worries by reducing the possibility of errors at each step of the process. In accordance, scientists from Granada, Spain, developed two pipelines for metagenomics and metatranscriptomics analysis and verified their performance through both simulated and experimental datasets.
PIPELINES
Metagenomic sequencing generates not the genetic makeup of a single isolate but a large pool of genetic data from all the microbial species present within that sample. This requires techniques that separate and classify the genomes of specific microbes to identify the population diversity within the sample. Amplicon sequencing targets specific rRNA sequences within microbes called the marker genes, shotgun sequencing that sequences the entire genome of the microbe, and metatranscriptomics, which studies microbial transcriptomic profiles, are the three major sequencing approaches for classifying the microbes, all with their own pros and cons. The pipelines described in the reference study can be adopted for any of the three methods.
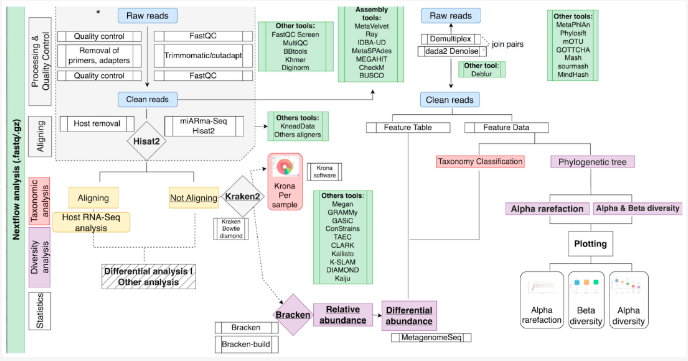
Image Source: https://doi.org/10.3390/genes13122280
The pipelines contain two main stages. The preprocessing and quality control stage involves filtering out low-quality reads, trimming the adapters, and removing the host reads and undesired sequences. The subsequent taxonomic classification stage involves the use of specific tools to compare the sequenced reads to reference sequences in a database to identify and classify the microbes from the sample data.
The choice between the two pipelines is based on the taxonomic classification step, which in retrospect is based on the choice of sequencing approach:
- Qiime Pipeline – Uses the QIIME2 tool to compare the marker genes sequenced from the sample with the reference database SILVA(database of rRNA sequences for marker gene alignment). It is designed to analyze amplicon-sequenced reads.
- Kraken-Bracken Pipeline – The Kraken2 tool uses the k-mers search (short read lengths that align perfectly with the reference sequence) to classify the read sequence whose abundance is estimated by the Bracken tool. The reference database used in this method is either SILVA or RefSeq(a curated version of GenBank containing databases of genomic DNA, transcripts, and proteins) based on the sequencing approach. The pipeline can handle amplicon, shotgun, and metatranscriptomics data.
EVALUATION OF PIPELINE PERFORMANCE
The authors assessed the pipeline performance on three datasets. The first two are synthetically generated microbiome data that are considered benchmarks for taxonomic classification to test the efficiency of the classifier. The last one is an experimental dataset on endometrial cancer that contains both marker gene and RNA-Seq data.
The first study using simulated metagenomic sequences contained three datasets of 10, 100, and 400 species, respectively. In the absence of rRNA data, the Kraken-Bracken pipeline with the RefSeq database was used. The pipeline calculated species abundances with high accuracy and marginal false-positive rates for the ten-species dataset. However, with increased species counts and reduced abundances, a higher rate of false positives was observed.
In the second study with synthetically generated gut microbiota data, both pipelines could be applied as the data provided was of 16S rRNA hypervariable region (marker genes of bacteria and archaea). In identifying organisms along the genus level, QIIME2 and SILVA produced better results by identifying all of the 58 genera present in the reference data, while Kraken2 with SILVA performed better in calculating the abundance of each genus. For species-level identification, it was noted that Kraken2 with SILVA did not identify a single species, while Kraken2 with RefSeq provided the best results.
Taking into account the results from the above two studies, both pipelines were run for the endometrial cancer study while excluding Kraken2-SILVA for identifying species within the sample data. For metatranscriptomic data from RNA-seq, the Kraken2-RefSeq pipeline was used, while QIIME2-SILVA was used for 16S rRNA data. The study identified genera and species of microbes in line with previously published literature on endometrial cancer.
Another important aspect to consider is the computational time and power required to execute the data analysis. The authors report the Kraken/Bracken pipeline to be 344 times faster and more accurate than the Qiime pipeline, though Qiime is recommended when the data is of 16S rRNA and the number of reads is low. With a large enough library, the authors conclude that at the genus level, Kraken2 provides more realistic results when combined with the SILVA database, while Kraken2 with the whole genome database, RefSeq, could provide better results at the species level with reduced computational cost and time.
CONCLUSIONS
In addition to the construction of the pipeline, the paper also summarizes the impacts of the microbiome on human health while providing an overview of the tools and techniques employed in analyzing the data generated through its sequencing. They further went on to address some of the major concerns one encounters when working with metagenomics data. Though this study discusses a few countermeasures, many still require newer technologies to resolve them. The authors hope to provide a guide to avoid at least a few of the pitfalls that could arise when tackling data as complex and as large as metagenomics data.
Article Source: Reference Paper | Pipeline Availability: GitHub
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Catherene Tomy is a consulting Content Writing Intern at the Centre of Bioinformatics Research and Technology (CBIRT). She has a master’s degree in Molecular Medicine from Amrita University with research experience in the fields of bioinformatics, cell biology, and molecular biology. She loves to pull apart complex concepts and weave a story around them.
.





{kind=link}