
Structure-based drug discovery (SBDD) is a standard method for identifying prospective medications for a target by leveraging its structural information. AlphaFold, a technique for predicting protein structure, has been regarded as a helpful resource for the discovery of therapeutics for new targets with low or no structural knowledge. In this study, the scientists utilized AlphaFold predictions as input for their AI-powered drug discovery engines (PandaOmics and Chemistry42) to efficiently identify a potential drug for CDK20 within 30 days.
Structure-based drug discovery (SBDD)
Understanding the structure of proteins is important in order to figure out their functions and the effect of change in amino acid sequence. The 3D structure of a protein allows us to visualize its functions and how genes and diseases are connected. This knowledge helps us make a decision about whether or not the protein would make a good target for drug creation.
Structure-based drug discovery (SBDD) is a popular method used for drug discovery. According to this, the 3D structure of a protein is examined and utilized in order to create smaller molecules that can bind to the protein and change its functions. Scientists aim to use SBDD to create medicines that would target the disease-causing proteins specifically, thus reducing the risk of harming the healthy ones.
Combining AlphaFold and AI
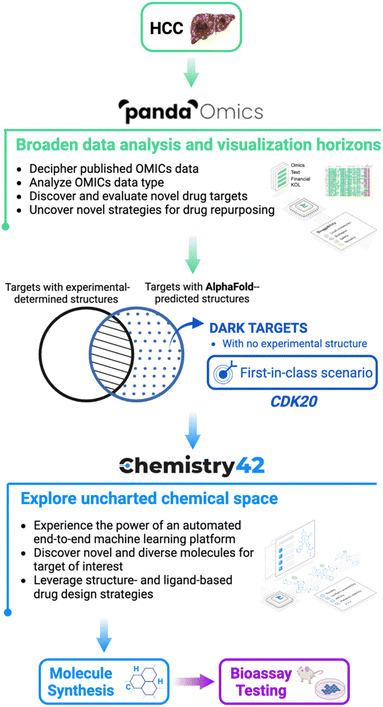
Image Source: https://doi.org/10.1039/D2SC05709C
AlphaFold is a revolutionary method for predicting the structure of proteins. It utilizes artificial intelligence to predict protein structures with an accuracy equivalent to that of experimental approaches. The introduction of the AlphaFold database, which contains over 800,000 protein structures, has expanded the scope of functional investigations, the identification of harmful mutations, and the investigation of protein interactions. AlphaFold’s influence has encouraged the development of other tools like RoseTTAFold and AlphaDesign, and it was recognized as a breakthrough of 2021 by the publications Science and Nature.
In this study, the scientists identified novel compounds for a particular target by combining AlphaFold, a protein structure prediction technology, with two AI-powered drug discovery platforms (PandaOmics and Chemistry42). Indication, target selection, hit generation, and hit recognition were all components of the procedure. As a starting point for their analysis, the scientists exploited the openly accessible predicted structures from the AlphaFold database. Even while AlphaFold is widely used in the scientific community, its use and adaptation for commercial applications still need to be understood.
AlphaFold, PandaOmics, and Chemistry42 were utilized to find new compounds for a novel target rapidly, cyclin-dependent kinase 20 (CDK20), which is linked to hepatocellular carcinoma (HCC) but has low experimental structural knowledge and few approved treatments. The authors analyzed text and OMICs data with PandaOmics and chose CDK20 as their target. Using Chemistry42 and AlphaFold’s anticipated CDK20 structure, they generated 8918 compounds, of which seven were chosen for synthesis and biological testing. The anticipated binding mechanism was employed for second-round compound synthesis and testing. This is the first instance of employing AlphaFold-predicted protein structures to effectively identify a verified hit for a new target in early drug discovery.
PandaOmics: choosing the best target to improve HCC treatment
Liver cancer, particularly Hepatocellular Carcinoma, is a commonly occurring and deadly disease. Unfortunately, treating this remains a challenge due to its high incidence and death rate. Despite this, recent studies have provided some hope. The combination of atezozilumab and bevacizumab has been proven to be a more effective first-line treatment option for people with advanced liver cancer compared to previous treatments, leading to improved overall survival and progression-free survival. Nevertheless, there is still a pressing need for better treatments for those affected by liver cancer.
PandaOmics is an AI-driven drug discovery platform that employs bioinformatics and deep learning to uncover possible therapeutic targets and biomarkers for a specific illness. It selects target genes using various scores generated from both text data and OMICs data and merges data from several studies into a single meta-analysis for exact target prioritization. In research for HCC, the platform provided a list of 20 prospective targets, and CDK20 was chosen as the most promising candidate based on its high scores and conformity to best-in-class standards. The target was then sent to the Chemistry42 platform, which generated small molecule inhibitors automatically.
Why CDK20?
CDK20 (cyclin-dependent kinase 20) is a protein found extensively in the human body, particularly in the digestive system, the brain, and the liver. It is also overexpressed in a wide variety of cancers, specifically lung cancer and liver cancer (Hepatocellular Carcinoma in particular). It is involved in a lot of processes, ranging from cell growth and survival to suppression of the immune system in a few tumors. A potentially useful treatment strategy that has emerged as per these findings is blocking CDK20, which may prove effective towards cancer, especially HCC.
There are currently limited CDK20 inhibitors identified in the scientific literature. One possible explanation for this is the absence of accessible 3D structural data for CDK20. Several CDK20 inhibitors, including RGB-286147, BMS357075, AAPK-25, Palbociclib, Flavopiridol, Dinaciclib, Roscovitine, and MER-128, have been reported. It has been reported that MER-128 is a strong CDK20 inhibitor with an IC50 value of 2 nM, although its structure is unknown. Through the Chemistry42 platform, CDK20 inhibitors are identified using a Structure-Based Drug Design (SBDD) strategy. The technology employs an energy-based strategy to identify potential binding sites by coating the protein surface with probes and estimating the energy of non-covalent interactions with the receptor atoms. Binding sites are discovered and graded using pocket volume, surface, and depth characteristics based on energy values.
Outcome
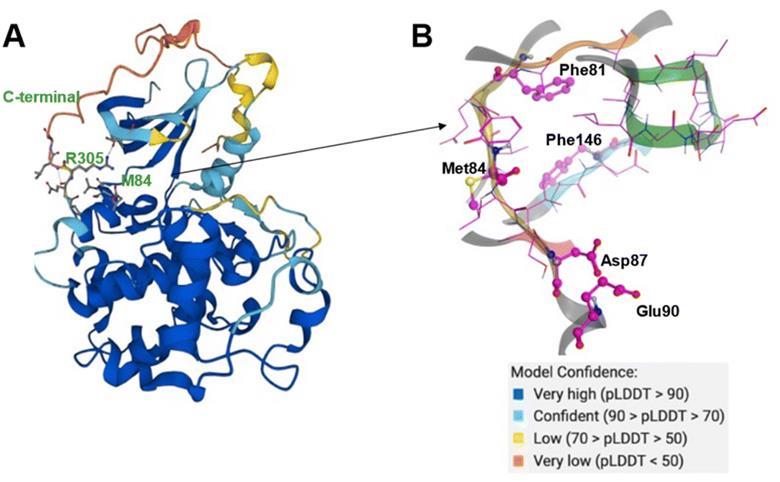
AlphaFold predicts the CDK20 structure with high confidence, with the exception of the C-terminal. AlphaFold predicts that the C-terminal of the protein blocks the solvent-exposed area and occupies the ATP pocket, rendering it unsuitable for the construction of an inhibitor. Therefore, only the structure from residue Met1 to Ile302 is employed to generate molecules in Chemistry42. The platform discovered a shallow ATP-binding pocket with an estimated volume of 150 A˚3 and a DFG-in conformation. The hinge residue Met84 is identified as the required binding site, and additional 3D structural information from the ATP pocket was used to direct the production of molecules. Chemistry42 created a total of 8918 molecules, of which 54 were prioritized, and 7 compounds were chosen for production.
The authors produced seven chemicals to evaluate their ability to bind to CDK20. One of the seven compounds, ISM042-2-001, exhibited binding affinity (Kd value) of 9.2 ± 0.5 mM and activity inhibition (IC50 value) of >6000 nM in CDK20 kinase binding and activity assays, respectively. This hit chemical was discovered in under 30 days. Through molecular docking, the scientists proposed the binding mechanism of ISM042-2-001 and discovered that it interacts with the hinge residue Met84, Leu85, and Ile10 via hydrogen bonds.
Using the AI tool Chemistry42, a second round of chemical production was done to increase the binding affinity of a CDK20 inhibitor. Six of the newly created compounds were produced and evaluated. Two of the compounds, ISM042-2-048 and ISM042-2-049, demonstrated an increase in binding affinity relative to the prior molecule. ISM042-2-048 was confirmed to inhibit CDK20 kinase activity with specific anti-proliferation activity in an HCC cell line with CDK20 overexpression and a novel scaffold. In the near future, more optimization and assessment of kinase selectivity, ADME (absorption, distribution, metabolism, and excretion) characteristics, and potency will be done.
Conclusion
SBDD is a popular technique for finding hit compounds and optimizing them as drug candidates. AlphaFold’s predicted protein structures are a vital tool for SBDD, particularly for new targets with low or no structural knowledge. In this study, the authors illustrate the quick identification of a CDK20 hit molecule by feeding predictions from AlphaFold into their automated drug discovery AI engines, PandaOmics and Chemistry42. This method, which included target selection, molecule formation, compound manufacturing, and biological testing, was concluded in thirty days. This research demonstrates the possibility of combining AI-powered drug discovery engines with AlphaFold predictions to find novel therapeutic targets and hit molecules rapidly.
Article Source: Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Sejal is a consulting scientific writing intern at CBIRT. She is an undergraduate student of the Department of Biotechnology at the Indian Institute of Technology, Kharagpur. She is an avid reader, and her logical and analytical skills are an asset to any research organization.





{kind=link}