
A recent study led under the supervision of Cenk Sahinalp et al. and conducted jointly at the National Institute of Health, UCSD, and Indiana University introduced an optimized framework for strain-level microbial identification. The team has provided a comparative approach with reference to other taxonomic classifiers and embedded results with higher sensitivity and specificity.
Introduction
Recent advancements in technology, such as High Throughput Sequencing (HTS) have eased the understanding of microbial diversity around humans. The isolation and identification of bacterial diversity range in different niches from environmental to human tissues. A decade of research has provided insights into the importance of these microbes in sustaining a healthy life, with their roles in causing and assisting in disease development. Tumorigenesis and the response to therapy have also been shown to have been impacted by the tissue-resident microbes. Considerably, several benchmark studies have prompted the use of these metagenomic HTS to develop taxonomic classification and abundance estimation methods.
The earlier alignment approaches for classification analyzed sequenced metagenomic data using reference databases such as GenBank through blastn or custom aligners such as GATK PathSeq. While HTS has subsequently provided efficient results in terms of accuracy, comparatively alignment-free approaches such as Kraken have been effective on large sample sizes. While alignment-based approaches primarily limit the classification to small samples with higher accuracy, the alignment-free approaches limit the classification by producing less accurate results.
The limitation associated with each approach such as the alignment-based approach can be facilitated by aligning the reads against a reference collection of sequences from marker genes which may limit the classification of low-abundance species due to selectivity based on marker genes. Similarly, the alignment-free approaches rely on an exact string or k-mer matching to grant taxonomic results for each read, wherein the approach differs in the accuracy of species-level or strain-level results. Hence, the method also deems insufficient for strain-level applications such as in order to identify mixed infections caused by multiple strains of a bacterial species.
Considering the earlier approaches and associated parameters, a team of researchers in collaboration with institutes such as NIH, UCSD, and Indiana University have introduced a computational approach to maintain and also manage the collection of bacterial genomes constructed in the form of strings /contigs representing bacterial species, a particular strain of the species or other taxonomic levels. This computational approach is CAMMiQ- Combinatorial Algorithms for Metagenomic Microbial Quantification.
CAMMiQ constructed a data structure that can answer questions in the following form: Given a set Q of HTS reads obtained from a mixture of genomes, each from S, identify the genomes in Q, and, in case the reads are genomic, compute their relative abundances. CAMMiQ uses an increased proportion of reads and can identify genomes error-free at sub-species and strain levels. The approach preferably has a more extensive selection of strings to choose from for selectivity in the sub-strings that increases the possible coverage. The team has conferred these results as theoretically based on the developed approach since these were not applied on the metagenomic datasets yet but are valid for CAMMiQ for the case c = 1 and other unique substring-based methods such as CLARK and KrakenUniq.
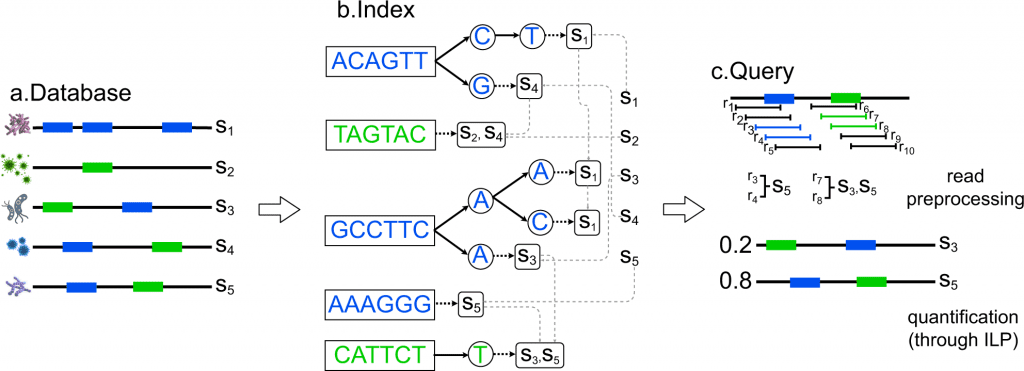
Image source: https://doi.org/10.1038/s41467-022-33869-7
The team believes the application of the following computational approach to single-cell data is of quite a relevance since studies surrounding commensal and pathogenic bacteria in the human microbiome can provide background in infections caused by microbes down to strain level. The current approach is more beneficial than single-cell nucleotide data and RNA sequencing approaches. Researchers have demonstrated a comparative analysis of CAMMiQ against the top-performing alignment-based and alignment-free metagenomic classification methods such as Kraken2, KrakenUniq, CLARK, Centrifuge, and Bracken on CAMI and IMMSA.
The methodology involved first introducing the overview of the CAMMiQ algorithm. CAMMiQ’s comparative accuracy performance against the alternative metagenomic analysis methods was based on the two species-level datasets: the first was the CAMI and IMMSA benchmark (i.e., species-level-all) index dataset and the second was the species-level-bacteria index dataset. On a surplus, the team has demonstrated the maximum advantage that the CAMMiQ could demonstrate through the use of double and unique, variable-length substrings on the species-level-bacteria index dataset.
The following overview was conducted by the team for comparative analysis:
1) The metagenomic classification or taxonomic profiling tool, CAMMiQ involves two steps, firstly the index construction and secondly the query.
2) The overall performance was based on four sets of experiments with distinct index datasets and distinct collections of queries. The comparison was based on the species-level-all dataset, species-level-bacteria index dataset, strain-level index dataset, and finally on the meta-transcriptomic reads dataset from a different study.
3) Species-level query for precision and recall in reading classification on CAMI and IMMSA. And performance evaluation of tools from benchmark studies.
4) Assessing the potential of variable-length and doubly-unique substrings in species-level-bacteria queries. According to its unique algorithmic features, CAMMiQ outperformed the available alternatives on the CAMI, IMMSA, and species-level-bacteria query sets.
5) The overall performance of the CAMMiQ tool outperformed other tools, the false readings were based more on the benchmark tools.
Final Discussion
The introduced novel computational tool CAMMiQ, to decipher the taxonomic classification of microbes from the high throughput sequencing data provide classification and identification to estimate the abundance of particular species or strain. The principal approach for the tool starts by defining the following algorithmic problem precisely that has not been fully addressed by the available methods. The approach dynamically improves the classification based on the use of doubly-unique sub-strings that increases CAMMiQ’s ability to identify and quantify the genome within the query.
Article Source: Reference Paper | Availability: CAMMiQ
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Mahi Sharma is a consulting Content Writing Intern at the Centre of Bioinformatics Research and Technology (CBIRT). She is a postgraduate with a Master's in Immunology degree from Amity University, Noida. She has interned at CSIR-Institute of Microbial Technology, working on human and environmental microbiomes. She actively promotes research on microcosmos and science communication through her personal blog.





{kind=link}