

Centuries after the first proteins were identified and characterized, their complexity proves a challenge for scientists all over the world. Their creation, evolution, and many variations are questions for which we have only just begun to uncover the answers. A new bioinformatics tool has now been developed that can provide researchers with a way to analyze data and map protein sequences and structures on a large scale. This has important implications for protein modeling and sequencing technologies.
Genomes provide the template on which all life is created and maintained. Despite the high accuracy of DNA replication mechanisms found in living organisms, certain mutations can still occur and be passed on. Such mutations can change the function and structure of the protein that the gene is supposed to encode. Nearly all Mendelian diseases are caused by variations in the proteome.
Current Issues in Protein Mapping
In lieu of the time-consuming and exhaustive process of determining protein structures and mutations in the lab, researchers have now turned to bioinformatics as an alternative method to quickly and cheaply generate highly accurate and precise protein structure models. Many protein modeling tools have been developed to predict mutations and their impact with as much accuracy and precision as possible. The majority of such tools use only protein sequencing information from both the protein as well as its variants in order to produce results. While such a method is advantageous in that it is applicable to nearly all genetic mutations, they don’t account for the structure of the mutated variants, resulting in a loss of valuable information that can be garnered from taking the proteins’ 3-dimensional structures into account. This is especially important as it allows for distinguishing between mere correlations in mutation occurrence and causative variations. Hence, the results obtained through such methods may not be as accurate or precise.
While some tools that utilize 3-dimensional structures are available, their utility is significantly hampered by the fact that experimentally derived structures are only available for a small portion of the proteins found in humans. Structural prediction of proteins, while it has advanced with the introduction of AlphaFold and similar technologies, is still in its infancy. Differences between sequences found in various databases like UniProt, Protein Data Bank, and Ensembl also add to the obstacles of accurately mapping 3-dimensional structural elements to their corresponding sequences, leading to difficulties in using bioinformatics tools. There may also be variations in the protein structures available in databases and those used in structural studies. Despite the progress in protein mapping technologies in recent years, many have become outdated due to a lack of updates and necessary modifications to algorithms as new discoveries regarding protein structure are made. Some tools are also specifically designed for certain organisms, rendering them useless for studies conducted on different organisms. In addition to these disadvantages, the majority of available tools are web-based. These tools tend to be a lot more accessible and convenient to users, but it also makes them unable to conduct data analysis on larger scales.
The Versatility and Power of 3Dmapper
3Dmapper circumvents these issues: it is a standalone tool that is able to map protein positions to the corresponding structures. 3Dmapper is very versatile and can be utilized for all organisms. If certain protein structures aren’t available, a sequence search is utilized, which can find relevant structural templates. Annotations of protein-ligand, protein-nucleic, and protein-protein interfaces are provided by determining distances between residues. In addition, 3Dmapper can work in tandem with the popular software AlphaFold 2, as it includes the B-factors for mapped residues, which correspond to pLDDT model quality metrics found in the models provided by AlphaFold 2. This also provides the user with crucial context and additional information.
Databases of structurally annotated proteins are generated when users input protein sequences or structures using BLAST software. Sequence identity cutoffs can be customized according to user requirements in order to maximize the number of structural homologs that are given. Inter-chain interfaces are then calculated through spatial proximity values provided by the user, and outputs are generated that contain detailed information about the given protein structures. Protein positions and variants can be mapped to a protein structure with the help of precomputed variants and structural files. 3Dmapper even includes functionality for visualizing the generated information, allowing users to visualize the mapped structures better.
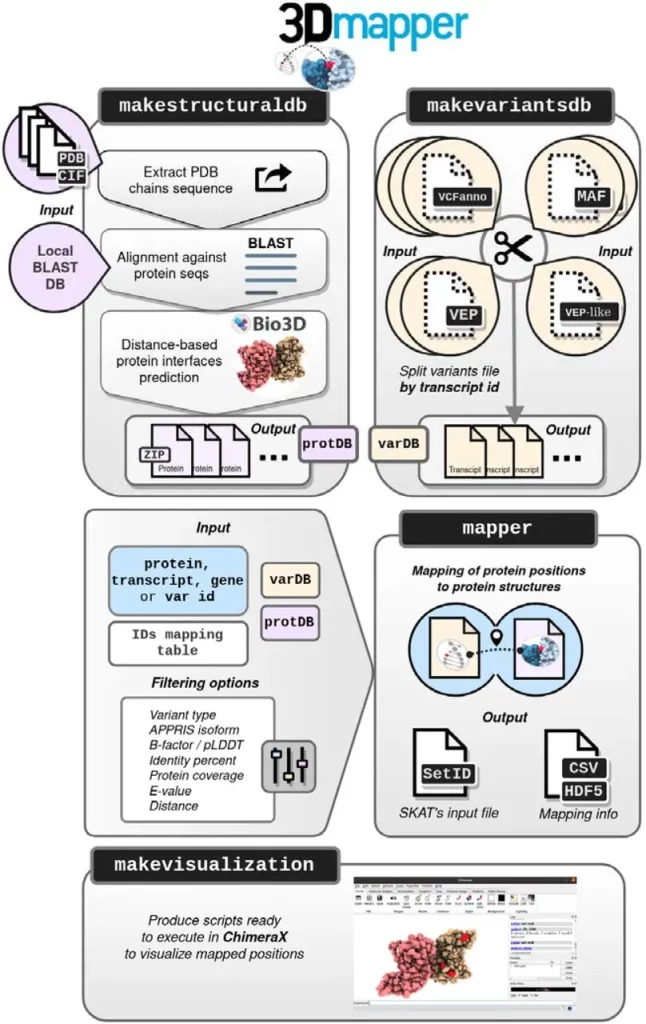
Image Source: https://doi.org/10.1101/2023.09.01.555502
Practical Applications of 3Dmapper
The performance and efficiency of 3Dmapper were tested to gain insights into its capacity for handling large-scale data analyses of variants from multiple databases, including ClinVar, TCGA, gnomAD, and the UK BioBank. The protein interactions predicted by 3Dmapper were evaluated and compared to interactions given by interactome3D, with 92% of all interactions being identified. More than 350,000 protein-protein interactions were also described, resulting in an increase in PPI structural coverage by 30 times. To gain a further understanding of the distribution of different mutations across various structural elements, repeated tests were performed for all four databases, with the variants being mapped to the structural database that had been previously generated. More than 19,000 transcript IDs were evaluated, with the final processing time being around 13 hours, which is reasonable for data analysis of genetic variants on such a large scale.
When various protein interfaces were evaluated, variants associated with disease were found to occur at a much higher number than variants that hadn’t been associated with any disease. These findings agree with prior studies, which concluded that missense variants were strongly enriched.
When missense variants were mapped to various structures and interfaces, the integration of high-quality AlphaFold models resulted in a 30% increase in the number of missense variants mapped in UK BioBank, gnomAD, and TCGA, with ClinVar also seeing an increase of 16%. To exemplify the utility of 3Dmapper when used alongside AlphaFold, mutations in XRCC2, a protein involved in DNA repair that is strongly associated with oncogenic mutations and variants, were studied. Experimental structures, as well as homology models, were unavailable for this protein interaction in PDB. Certain mutations inside this protein were predicted to change the protein’s ability to function, making these variants potentially significant for further clinical research.
In addition, 3Dmapper (and protein mapping) can also be used for many other applications, like analyzing somatic mutations and the potential consequences caused by certain variations or identifying protein positions of evolutionary significance.
Conclusion
3Dmapper thus serves as a way to further our comprehension of the mysteries of protein structures as well as protein modeling. With multiple potential applications in the fields of biotechnology, bioinformatics, and medical technology, 3Dmapper will allow researchers to perform genomic studies and protein mapping on a much larger scale than ever before.
Article Source: Reference Article | 3Dmapper is available freely on GitHub
Learn More:




Sonal Keni is a consulting scientific writing intern at CBIRT. She is pursuing a BTech in Biotechnology from the Manipal Institute of Technology. Her academic journey has been driven by a profound fascination for the intricate world of biology, and she is particularly drawn to computational biology and oncology. She also enjoys reading and painting in her free time.





{kind=link}