
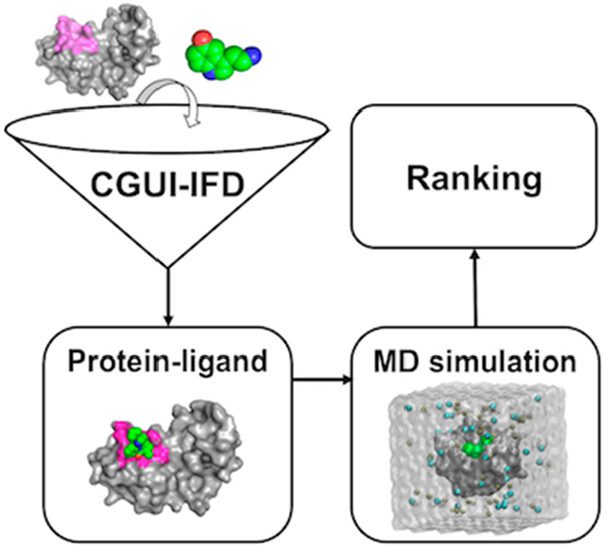
Researchers from Leigh University, United States, have developed a novel technique referred to as CHARMM-GUI protein-ligand docking (CGUI-IFD) to overcome the drawbacks associated with molecular docking in drug design. Molecular docking strives to comprehend how a potential pharmaceutical compound (ligand) associates with a distinct protein (receptor) in an organism. This method helps researchers determine the binding affinity and potential effectiveness of a drug candidate and also assists scientists in forecasting the extent to which the medication will attach to the protein structure. Nevertheless, a challenge exists during this procedure due to the protein’s potential to transform its shape when the drug binds to it. This is referred to as the “induced-fit effect,” making docking complicated. CGUI-IFD is a sequential method that enhances the protein’s interaction site, connects the medicine to the target, and simulates the way they engage in a dynamic environment. Scientists evaluated the approach using a collection of various proteins and pharmaceuticals. A study revealed that the method accurately forecasted the proper binding configurations in 80% of the cases.
A Barrier to Efficient Docking: The Induced Fit Problem
In drug design, a correct understanding of how a drug particle interacts with a particular protein is essential. The understanding is obtained via protein-ligand complex structures. Such arrangements demonstrate the specific interplays between the pharmaceutical and the protein’s adherence spot. Experimental methodologies such as X-ray crystallography and cryo-EM are regarded as the epitome for uncovering these structures. However, these strategies are usually expensive and time-intensive. This makes it challenging to use them for every new drug being studied. To address the drawback, experts suggest an algorithmic approach that can swiftly and economically anticipate how the medication and protein will engage, creating reliable protein-ligand interaction modes.
Over one hundred years ago, scientist Emil Fischer developed the lock-and-key theory to clarify the connection between proteins and drugs interacting with each other. That concept continues to be broadly implemented currently within the realm of biochemistry. He visualized the molecule’s active site similar to a padlock. The drug candidate, on the other hand, was viewed as an exact key that fits seamlessly in it. This concept has been utilized in numerous computational drug design methodologies to estimate how drugs attach to protein molecules. These methods consider that the amino acid’s form stays unchanged, similar to a solid framework, and look in pursuit of the drug molecule that satisfies the criterion.
Nonetheless, this strategy has its drawbacks owing to the overlooked occurrence of “induced fit.” Induced fit takes place when the protein is capable of altering its shape slightly to bind a specific drug better. This leads to the conventional lock-and-key becoming less accurate. Researchers had recognized the concept of induced fit since 1958 when it was initially introduced by Koshland. Scientists now understand that molecular binding sites are flexible structures that have the ability to take on different configurations depending on the drug they bind to. It makes it harder for computational approaches to estimate the exact interplay between proteins and substances.
Scientists have developed various computational methods to tackle the induced fit docking (IFD) problem, including Schrodinger IFD-MD, CIFDock, Fleksy, and tinyIFD. These methods aim to predict the complex interactions between drugs and proteins, taking into account the protein’s ability to change its shape slightly to accommodate the drug better. Schrodinger IFD-MD, for example, combines several steps, like molecular dynamics simulations, to generate protein conformations, rigid docking, and energy-guided structure modeling. Similarly, CIFDock employs Langevin dynamics simulations to sample relevant drug conformations and protein movements. Fleksy and tinyIFD use simpler approaches but still consider protein flexibility during docking.
The most common method, however, is the ensemble MD-docking approach, which involves using multiple protein structures obtained from molecular dynamics simulations and performing rigid docking on each to account for induced fit. While these methods show promise, challenges remain, particularly in accurately identifying the correct protein structures for docking in some cases.
CHARMM-GUI: The Antidote to the IFD Problem
Researchers Hugo Guterres and Wonpil Im present a new and straightforward method called CHARMM-GUI-based IFD (CGUI-IFD) to study how drugs interact with proteins. Unlike Schrodinger IFD-MD, which relies on proprietary technologies not accessible to most researchers, and ensemble MD-docking, which has limitations in identifying accurate protein shapes from clustering, CGUI-IFD uses the widely available CHARMM-GUI web-based tool for preparing molecular simulation systems. The workflow consists of two main steps: LBS-FR (ligand-binding-site Finder & Refiner) and HTS (High-Throughput Simulator). LBS-FR refines the protein’s binding site to create different shapes the drug can fit into, while HTS performs docking of the drug into these different protein structures. The best binding mode is selected based on stability (measured by ligand root-mean-square deviation, RMSD) and how well it interacts with the protein (using molecular mechanics with generalized Born surface area solvation). The method was tested on 258 protein-drug structures and successfully predicted the correct binding mode in 80% of cases, demonstrating its reliability and user-friendliness for studying drug-protein interactions in drug discovery research.
Witnessing the Brilliance of CHARMM-GUI
CGUI-IFD has a straightforward workflow
The CGUI-IFD workflow is quite uncomplicated. It begins by refining the protein’s binding site using a library of templates from experimentally solved protein-ligand structures. This generates an ensemble of four receptor-binding site structures, including the original one and three refined ones from the templates. Ligands are then docked into these structures using rigid docking, resulting in 40 different protein-ligand binding poses for each case. The ligand-binding poses are processed using the High-Throughput Simulator (HTS), which evaluates their stability through 50 ns molecular dynamics (MD) simulations. Stable poses with ligand root-mean-square deviation (RMSD) less than 3 Å are selected. The final structures are ranked based on their interaction energy using the molecular mechanics generalized Born surface area (MMGBSA) method.
CGUI-IFD achieves 80% accuracy in generating protein-ligand binding modes
The researchers conducted a benchmark test using a dataset of 258 protein-ligand pairs to validate their CGUI-IFD method. Impressively, CGUI-IFD achieved an 80% success rate in accurately predicting the correct protein-ligand binding modes in these test cases. They found that using LBS-FR to generate additional receptor structures significantly improved success rates, highlighting its importance in accounting for induced fit effects. Comparing CGUI-IFD to other methods, it outperformed rigid receptor docking approaches and showed similar performance to another method called tinyIFD. As an illustrative example, a case involving mitogen-activated protein kinase-2 (MK-2) demonstrated that CGUI-IFD accurately predicted the correct ligand-binding mode, effectively overcoming clashes observed in rigid docking. Overall, CGUI-IFD generated an ensemble of 40 protein-ligand structures, and the top-scoring structure had a binding affinity of -25.1 kcal/mol. These findings emphasize the effectiveness of CGUI-IFD in predicting reliable protein-ligand interactions, especially considering induced fit effects, and showcasing its superiority over traditional rigid docking methods.
CGUI-IFD even transforms many failed cases into success with an increase in template number
CGUI-IFD, a powerful method for predicting protein-ligand binding modes, excels even in cases with induced fit effects. In initial tests, using three template protein conformations produced favorable results, achieving an 80% success rate. Seeking to improve further and transform the rest 20% of the cases into success, researchers increased the number of templates to 10 and tested the approach on ten diverse cases from different protein target classes. Remarkably, CGUI-IFD successfully rescued and accurately predicted the correct ligand-binding mode in 8 out of the 10 cases.
A Look at the Limitations of CGUI-IFD
CGUI-IFD, while successful in predicting reliable protein-ligand binding modes, encountered persistent challenges in two cases. In the first case, involving the protease coagulation factor VIIa, the ligand’s intricate structure with numerous rotatable bonds caused solvent-exposed groups to flip, resulting in inaccurate binding predictions. The second case, a cross-docking scenario between receptor Hsp70 and ligand 3F5, presented complexities due to a delicate hydrogen bond network forming a narrow binding pocket. Consequently, CGUI-IFD struggled to accommodate the ligand’s adenosine group correctly within the pocket. These challenges shed light on the intricacies of ligand structures and hydrogen bonding, necessitating future improvements in protein-ligand docking methods.
Conclusion
Molecular docking is highly essential to drug designing, but its efficiency gets hindered by the induced fit problem. CGUI-IFD tackles the challenges of induced-fit docking by combining two CHARMM-GUI modules to generate a variety of receptor conformations through MD simulations with template restraints. The workflow identifies the appropriate candidate poses based on ligand RMSD and MMGBSA binding energy by rigid docking and high-throughput explicit solvent MD simulations. Successfully validated with an 80% success rate using a large data set of 258 cross-docking cases, CGUI-IFD offers an alternative method for academic researchers, providing reliable protein-ligand structures without the need for expensive experimental determination. Notably, CGUI-IFD is not restricted to holoprotein receptors and works with apo and homology model structures. The versatility extends to supporting various MD simulation programs and multiple ligand force fields for efficient parametrization. Even though it has minor limitations, this robust and cost-effective approach holds significant potential for enhancing structure-based drug discovery by delivering accurate binding modes for a wide range of compounds.
Story Source: Reference Paper
Learn More:




Neegar is a consulting scientific content writing intern at CBIRT. She's a final-year student pursuing a B.Tech in Biotechnology at Odisha University of Technology and Research. Neegar's enthusiasm is sparked by the dynamic and interdisciplinary aspects of bioinformatics. She possesses a remarkable ability to elucidate intricate concepts using accessible language. Consequently, she aspires to amalgamate her proficiency in bioinformatics with her passion for writing, aiming to convey pioneering breakthroughs and innovations in the field of bioinformatics in a comprehensible manner to a wide audience.





 for reliable binding mode predictions.){kind=link}