
Substantial progress has been made in assembling the first complete human genome sequence by the Telomere-to-Telomere consortium. However, complex repetitions in diploid genomes are still difficult to decode. Using lengthy, accurate reads and haplotype-specific markers, the Verkko pipeline is an automated method for assembling diploid genomes with high precision. The construction of pangenomes and chromosome-scale comparative genomics using Verkko would be particularly useful in the field of evolutionary biology.
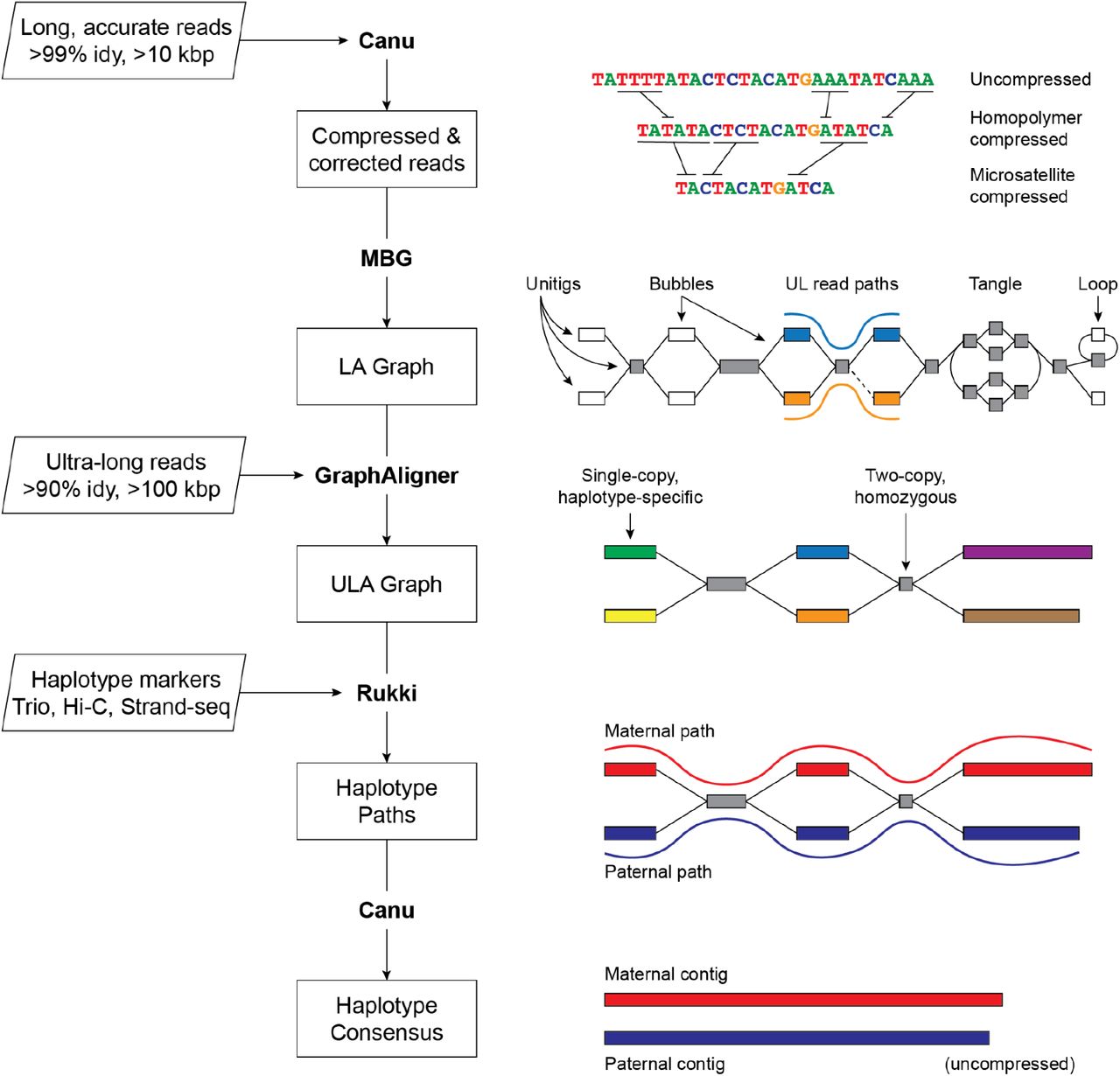
Combining Long, accurate and Ultra-long reads
The increased accuracy and length of sequencing reads made it simpler to rebuild genomes from overlapping reads as a result of technological advances in sequencing. Long, accurate reads and ultra-long reads have made it possible to yield extremely continuous genomic assemblies. Recent work has proved the viability of gapless and efficient assembly of human genomes; nevertheless, it still needs substantial resources and is not yet a common procedure. The chromosome-scale structuring of LA-based assemblies necessitates the use of other technologies, like Bionano, Strand-seq, or Hi-C, which are error-prone and need meticulous curation and validation. Despite these obstacles, advancements in sequencing technology have significantly enhanced the capacity to rebuild genomes and have the potential to advance genetic research.
Very similar repetitions make it hard to put together the genome. Long, accurate (LA) reads are good at resolving diverged repeats but not exact repeats. Ultra-long (UL) reads, on the other hand, can cover exact repeats but aren’t as accurate. Verkko is a new assembler that uses both LA and UL reads to get rid of repeats. To put together diploid chromosomes, it uses haplotype information from familial trios, Hi-C, or Strand-seq. Verkko’s iterative pipeline builds a multiplex de Bruijn graph, combines UL reads and haplotype-specific markers, and improves the assembly over time. Verkko can put together diploid genomes with high accuracy. This makes it possible to build complete pangenome databases and does comparative genomics at the chromosome scale.
High-resolution Assembly Graph
Verkko constructs a high-resolution assembly graph from long-read sequencing data using a graph-first technique. The approach is intended for use with diploid genomes and is entirely automated. The process runs on homopolymer-compressed sequences and utilizes LA reads to construct a multiplex de Bruijn graph, which is resolved further by UL reads and haplotype markers. The final network is cleaned, and haplotype pathways are found in order to restore homopolymers, which leads to the computation of consensus sequences for all nodes and haplotype paths. The approach resolves divergent repeats and catches minor mutations between haplotypes, resulting in a diploid genome assembly of superior quality.
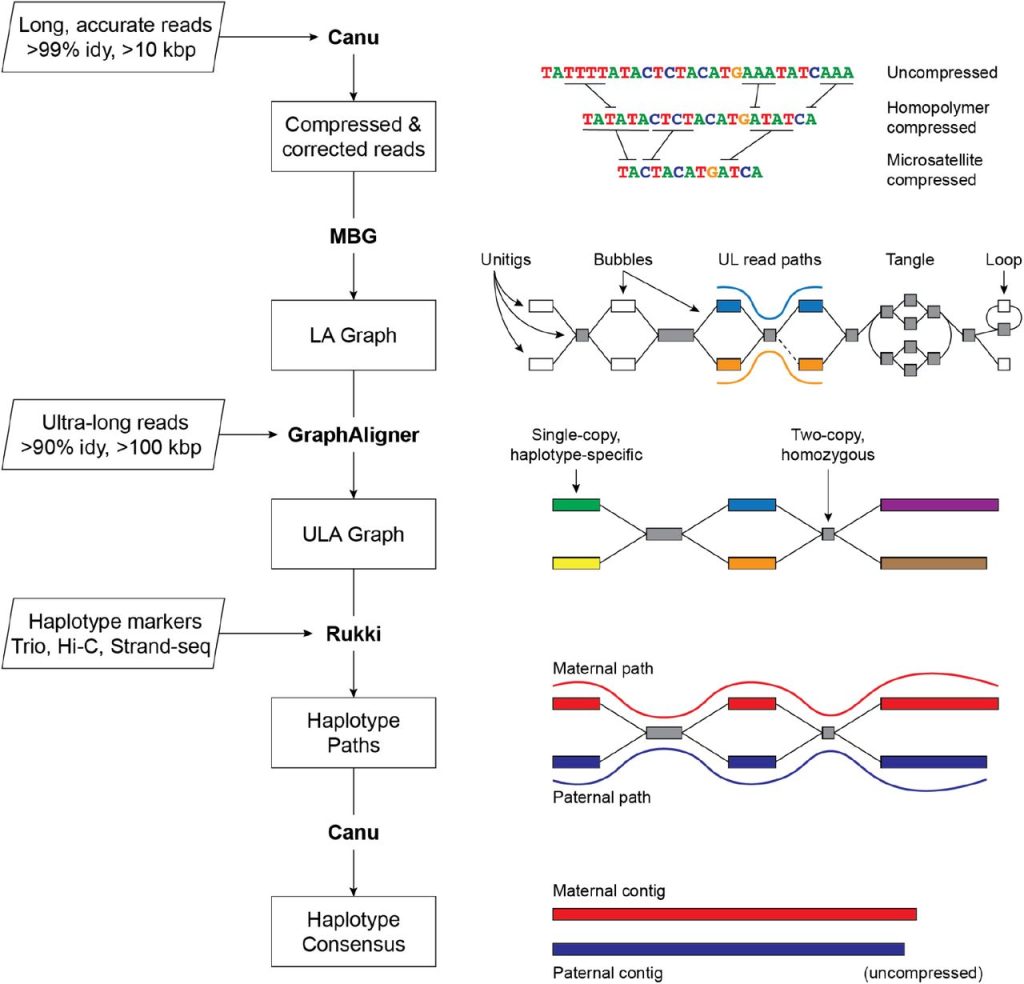
Image Source: https://doi.org/10.1038/s41587-023-01662-6
The end output of Verkko is a phased, diploid genome assembly that distinguishes maternal and paternal haplotypes, resulting in a highly accurate and resolved assembly graph. In certain instances, the given data may not be adequate to determine a chromosome as a single contig. Verkko utilizes the assembly graph structure in such instances to build haplotype-resolved telomere-to-telomere scaffolds. The assembly graph depicts the relationships between the various parts of the genome, including areas that may be difficult to assemble due to high divergence or repeating sequences. By utilizing the graph structure to produce scaffolds, Verkko avoids the requirement for a separate, error-prone scaffolding phase required by many other genome assembly techniques. This results in a more precise and continuous genome assembly that may be utilized for further analysis and genome completion.
Testing the Verkko model
The Arabidopsis thaliana genome and the CHM13 human genome were used to test the Verkko genome assembler. The study compares Verkko to other cutting-edge genome assemblers intended for HiFi and ONT data, using a variety of assessment measures such as assembly completeness, accuracy, and correctness.
Using HiFi and ONT data, Verkko was able to assemble 4 out of 5 chromosomes of Arabidopsis thaliana into single unitigs spanning >99.5% of the reference genome with the lowest error rate and equivalent base accuracy to other HiFi-based assemblers. Verkko was similar to other HiFi-only assemblers when utilizing only HiFi data, however, it failed to resolve any chromosomes end-to-end.
Verkko correctly assembled 12 chromosomes from telomere to telomere in the CHM13 human genome, with 5 extra chromosomes integrated into a single unitig having >95% of the predicted sequence. This is double that of any assembly based on a single technology, with the La Jolla Assembler (LJA) attaining the next greatest achievement by assembling six chromosomes using HiFi data alone.
Assembling complex regions
While the complex regions, including the rDNA arrays and centromeric satellite arrays, were not completely resolved, Verkko was able to partly separate the chromosomes and accurately identify missing components in other places.
The ability of Verkko to mix multiple data types (LA and UL) proved useful in resolving complicated areas, such as the GA-rich microsatellite on Chr8 that needed ONT-based gap-filling to be properly resolved.
Verkko was able to build the highly repetitive and complicated portions of the genome with greater precision and completeness than other genome assemblers. In the case of ChrX, for instance, previous assemblers required numerous contigs to cover >99% of the genome, but Verkko was able to construct a single unitig that covered >99.98% of the genome, with the final 50 kb on the q-arm missing due to a retained heterozygous bubble.
The VerityMap output for the Verkko assembly was personally examined and found to be in better agreement with the CHM13 reference in key locations, indicating that certain reported QUAST problems may be false positives or mistakes in the reference genome.
Diploid Genome Assembly
Verkko was assessed further on two distinct genomes: the highly heterozygous Darwin Tree of Life insect genome and the HG002 human sample used as the standard. The researchers compared Verkko’s output of phased unitigs to completely phased counterparts from other assemblers and discovered that, despite very limited ONT data, Verkko’s unitig NG50 was comparable to the pseudo-haplotype reference and significantly bigger than other assemblers. In addition, they discovered that Verkko’s combination of HiFi and ONT sequencing yielded very accurate and comprehensive assemblies that were comparable to the best unphased pseudo-haplotypes for various genomes.
With the addition of Hi-C data, Verkko’s contig NG50 rose even more, surpassing the pseudo-haplotype reference’s continuity. Verkko was then applied to the benchmark human sample HG002 utilizing a downsampled 35x dataset base-called using DeepConsensus with 60x ONT ultra-long reads. They compared the outcomes with the recently completed HG002 ChrX and ChrY, utilizing QUAST and measuring assembly quality and precision with reference-free approaches.
Comparison with Hifiasm and other assemblies
Verkko and Hifiasm assemblies are complete, with Verkko recovering somewhat more multi-copy genes at the price of a slightly greater proportion of spurious duplications within particular haplotypes. Verkko is able to create correct scaffolds using only the connectivity information included in the assembly graph, generating chromosome-scale scaffolds for seven and four chromosomes with trio and Hi-C information, respectively, and T2T scaffolds with much lower HiFi and ONT coverage. This feature prevents a separate, error-prone scaffolding phase. Nevertheless, repeated sequences in Verkko can be handled using postprocessing techniques such as purge dups or enhanced Hi-C handling in the absence of trio information.
The study used the HG002 dataset with complete coverage to generate the most continuous assembly of this genome to date. A comparison was made between the Verkko assembly, a Hifiasm triple assembly, and a recently released benchmark assembly. The Verkko trio and Hi-C assemblies have fewer faults and more precise phasing than the other assemblies. Verkko was able to decipher intricate sections of centromeric repeat arrays, allowing for more precise phasing. Using triple information, Verkko resolves 27 of the 46 chromosomes in HG002 into single scaffolds, 20 of which are constructed from telomere to telomere without gaps, a considerable improvement over prior assemblies. Verkko was able to resolve nine chromosomes into single scaffolds, seven of which are gapless, using only Hi-C data.
Conclusion
Verkko is a new assembler that uses both LA and UL reads to make genome assemblies that are more accurate and continuous. It uses high-accuracy reads to build an initial assembly graph, which is then refined with long, noisy reads. Haplotype markers are mapped to find haplotype paths through the graph. Verkko doesn’t work with ONT’s “simplex” sequencing, and it takes longer to run than some assemblers that only work with HiFi. Verkko currently recommends sequencing about 50x coverage of LA reads, 50x coverage of UL reads >100 kb, and 50x coverage of parental short reads for a complete diploid genome assembly. Verkko is modular and can be used with different technologies, but its heuristics assume uniform coverage, so it may not be the best choice for assembling metagenomic data without more work.
Article Source: Reference Paper
Learn More:




Sejal is a consulting scientific writing intern at CBIRT. She is an undergraduate student of the Department of Biotechnology at the Indian Institute of Technology, Kharagpur. She is an avid reader, and her logical and analytical skills are an asset to any research organization.





{kind=link}