
Are you aware that plenty of operational AI applications have emerged in the early stages of small-molecule drug discovery? Do you recognize that those packages regularly require a thorough grasp of software programs and hardware, as well as a specialization in an optimistic effort, inflicting boundaries for regular users, in fact, normalization and switching among them? Researchers at Zhejiang Shuren University and Hangzhou SanOmics Information Technology created MolProphet, a user-friendly software with an AI-based total interface to cope with this impediment. MolProphet offers a complete answer for small molecule drug development, together with AI-pushed target pocket prediction, hit discovery, lead optimization, compound concentration, and powerful analytics tools. Its design seeks to simplify the process, permitting ordinary people to navigate through the numerous tiers of drug research.
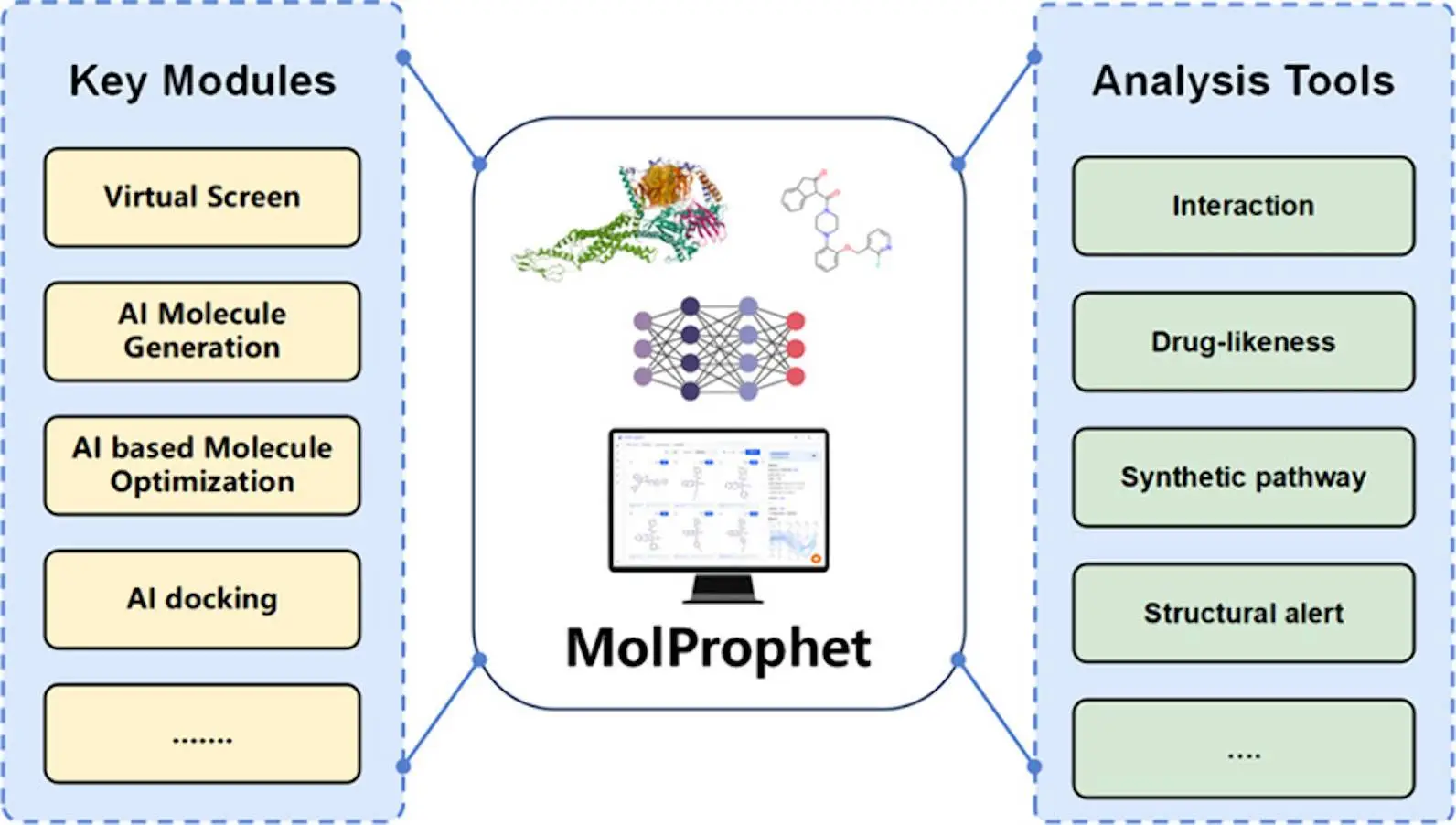
Crucial Mechanisms: Key Modules Supporting the Research
Pocket Discovery and Management Component: This module facilitates the discovery and control of likely interplay pockets on proteins, which can be critical for drug enhancement. It makes use of deep perusing techniques to antedate modest pocket edifices automatically. Moreover, it allows the manual definition of pocket topologies, resulting in associated profiles for every pocket.
The Structural Component: It uses a deep neural community trained on massive amounts of bioassay input to speed up the virtual screening of commercially available chemical libraries, which vividly enhances screening performance with the aid of screening hundreds of thousands of molecules in a quick length.
Ligand-centric Method: It uses AI approaches to extract molecules similar to recognized drugs from commercial libraries swiftly. It makes use of 2D-3D structure similarity strategies to repossess millions or even billions of molecular information quickly.
AI-driven Generative Component: It prioritizes drug-likeness, synthesis feasibility, and charge effectiveness to create innovative, profitable, and simply synthesized compounds. AI helps to construct compounds from quite simply available building additives and gives synthesis strategies for each generated molecule.
Molecule Optimization Components: These segments offer plenty of ways to optimize molecules, including scaffold hopping, fragment optimization, and R-group optimization. Users can choose the most suitable optimization approach for their drives, which includes optimizing the whole molecule or deciding on sections.
AI Docking Module: This module makes it simpler to estimate molecular datasets rapidly using artificial intelligence technology. It uses geometric deep learning and reinforcement studying to estimate the minimum free electricity of molecules binding to particular pockets, which helps with chemical screening and assessment.
Scrutiny of Toolkits: Renovating Raw Data into Valuable Insights
Oral Bioavailability & Structural Caution Evaluation: The platform analyzes materials using Lipinski’s rule of five, which takes into account physicochemical aspects consisting of molecular weight (MW), topological polar surface area (TPSA), lipophilicity (logP), hydrogen bond donors (HBD) & acceptors (HBA). This valuation enables regulation of the compound’s viable oral bioavailability.
Structural cautions are purposeful consortia or sub-structures that can be associated with toxicity or low bioavailability. The platform includes regulations for identifying and addressing structural alarms at some stage in the design phase, such as the BMS Rule, Chelator Rule, PAINS Rule, Genotoxic Carcinogenicity Rule, NTD Rule, and SureChEMBL Rule. This allows for the dealing of protection and effectiveness troubles, decreasing the chance of late-degree turnover in medicinal research.
ADMET Prediction: Understanding ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) is vital for determining drug safety and effectiveness. The platform evaluates absorption, bioavailability, metabolism, excretion, and toxicity using strategies including Caco2, HIA, P-gp Inhibitor, Hydration Free Energy, BBB Penetration, Plasma Protein Binding Ratio, CYP Inhibitors/Substrates, Half-Life, Clearance, and toxicity indicators including hERG, DILI, Carcinogenicity, LD50, Respiratory toxicity, and Eye Corrosion. These reviews help assume drug behavior earlier than clinical trials start, improve patient well-being, and lower the prospect of medication failure.
Binding Mode Evaluation: The platform includes techniques for scrutinizing interactions between molecules and ligands. This contains 3D visualization of protein-ligand complexes, binding mode analysis that shows interactions primarily based on pressure type, and binding site evaluation that statistically analyzes in all likelihood ligand contours. Future upgrades to this module might consist of dangerous substructure suggestions and proprietary molecular reviews.
Real-World Scenarios: Practical Applications Unveiled
Since its release in 2017, MolProphet has been actively used in over 20 hit discovery and lead optimization projects, with mind-blowing consequences. As of December 10, 2023, the Software as a Service (SaaS) platform had contributed to over six hundred initiatives as a consequence of its increasing person base of over four hundred people. Notably, MolProphet has excelled in finding molecules to the micromolar degree after optimizing them to acquire nanomolar potency.
Concluding Comments
MolProphet is a smooth-to-use, AI-powered platform for early-level small-molecule drug improvement. Its relation with AI expertise permits researchers to easily harness the software without requiring substantial computational expertise. The platform gives virtual screening, molecule design, and optimization skills, in addition to proprietary ways for green chemical synthesis. MolProphet permits lead optimization through scaffold hopping, fragment optimization, and R-group. Overall, it is a useful internet resource for researchers in the early levels of drug discovery.
Article source: Reference Paper | MolProphet is available for everyone on the website.
Follow Us!
Learn More:




Anshika is a consulting scientific writing intern at CBIRT with a strong passion for drug discovery and design. Currently pursuing a BTech in Biotechnology, she endeavors to unite her proficiency in technology with her biological aspirations. Anshika is deeply interested in structural bioinformatics and computational biology. She is committed to simplifying complex scientific concepts, ensuring they are understandable to a wide range of audiences through her writing.











{kind=link}