
Scientists from the Hengyang Normal University, China, designed a stacked autoencoder (SAE) with a multi-head attention mechanism-based microbe drug association prediction model named MDASAE. Intensive comparison experiments and case studies based on public databases under 5-fold cross-validation and 10-fold cross-validation, respectively, proved the superiority of MDASAE to existing other calculation methods, indicating its prospects in acknowledging microbe drug association, novel drug discovery, and effectiveness in clinical treatments as well.
The Human Microbiome: Implications for Health, Disease, and Personalized Medicine
Humans and the microbiome share a mutually constraining and interdependent relationship. The presence of a microbial community within us is essential for maintaining a state of balance, but it also carries the risk of diverse complications. These complications include atherosclerosis, inflammatory bowel diseases, alcoholic liver disease, metabolic syndrome, diabetes mellitus, cancer, obesity, cardiovascular disease, and hypertension, among others. Evidence of microbiota implying pharmacodynamics, pharmacokinetics, bioavailability, metabolism, safety, and effects of therapeutics synergistically or antagonistically is bringing another dimension to the establishment of personalized medicine. Microbes can be a potential drug target. Therefore, comprehending drug-microbe interactions becomes essential for drug development, drug repurposing, screening, and disease diagnosis.
Additionally, the emergence of drug resistance demands more meticulous studies in various aspects. However, conventional wet lab experiments are wasteful of money, time, and resources. Computational tools developed by integrating available datasets through machine learning complement this limitation, making drug discovery feasible and faster.
Microbe-Drug Association Database (MDAD)
The database MDAD is a collection of 5,055 entries of clinically and experimentally supported data of microbe-drug cognates, including 1,388 drugs and 180 microbes from multiple drug databases and related publications, with detailed annotations for each record, such as hyperlinks from DrugBank, Uniprot, and the original reference links ‘aBiofilm,’ the database that contains biological, chemical, and structural details of 5027 anti-biofilm agents reported from 1988–2017.
The MDASAE Model
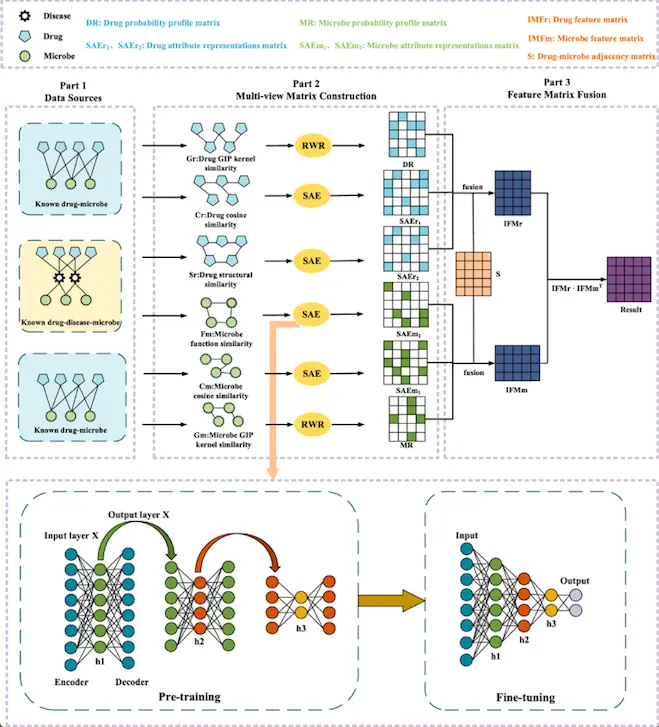
Based on the downloaded known microbe-drug association data from MDAD and aBiofilm, the scientists built a microbe-drug adjacency matrix that differentiates between possible drug-microbe association and no association. The drug similarity network and microbe similarity network are constructed following Gaussian kernel similarity. For the drug similarity network, chemical structure similarity scores were calculated using SIMCOMP2. The Restart Random Walk (RWR) algorithm is applied in the derivation of inter-node features for microbes and drugs, obtaining a drug probability profile matrix and a microbe probability profile matrix.
A stacked autoencoder (SAE) is first pre-trained in an unsupervised manner, and then a supervised method is employed to fine-tune the parameters to learn the attribute features between nodes. SAE could learn the most important attributes of the input data and reconstruct the input data in the output layer through encoding (mapping the input data to the hidden layer) and decoding (mapping the hidden layer to the output layer). A multi-head attention mechanism into the output layer SAE is introduced to capture critical features and improve the efficiency and accuracy of the attribute feature extraction process. The introduction of the Adam optimizer for training in SAE resulted in two different drug attribute feature matrices and two different microbe attribute feature matrices. Microbes- and drug-related node attribute features and inter-node features are fused to estimate possible association scores of different microbe-drug pairs.
Gaussian kernel similarity is used in machine learning and pattern recognition to calculate the similarity between two data points depending on their distance in a high-dimensional feature space. SIMCOMP (SIMilar COMPound) is a graph-based method for comparing chemical structures, which searches for the maximal cliques in the association graph as the maximum common induced subgraph (MCIS). Restart Random Walk algorithm allows the discovery of different patterns or properties of the large graph or network.
A Stacked autoencoder (SCA) is a neural network consist several layers of sparse autoencoders for unsupervised learning, dimensionality reduction, and feature extraction, where the output of a hidden layer is connected to the input of the next hidden layer. The multi-head attention mechanism captures different types of relationships or patterns in the input sequence by parallel processing local and global dependencies, facilitating better information integration. The Adam optimizer is an adaptive optimization algorithm utilized in training deep learning models to handle the learning rate for each parameter.
The effects of relevant parameters on the predictive performance of MDASAE for model optimization are analyzed. They evaluated the effect of the learning rate and the number of attention mechanism heads on the prediction performance of MDASAE while its value varies in different ranges. Predictive performance between MDASAE (with the multi-head attention mechanism) and MDASAE W/O attention (without the multi-head attention mechanism) was done. All of these experiments showed favorable outcomes.
Moreover, MDASAE outperformed representative state-of-the-art competing calculation models [LRLSHMDA30 (Laplace regularized least squares classifier, a semi-supervised to predict potential microbe-disease associations), HMDAKATZ12 (KATZ measure-based calculation method to infer latent associations between microorganisms and drugs) BIRWMP31 (multi-path based bi-random walk to detect possible microbe-disease associations), NTSHMDA32 (random walk algorithm to infer potential microbe-disease associations by integrating network topological similarity) andLAGCN33 (embeddings from multiple graph convolutional layers with an attention mechanism to predict latent microbe-disease associations)]. A case study was performed with two drugs- Pefoxacin and Ciprofloxacin and microbe Mycobacterium tuberculosis. The model is able to predict results that can be confirmed by published reports.
Conclusion
The researchers expect MDASAE to be employed for other association prediction issues between biological entities, such as microbe-disease association prediction and circRNA-disease association prediction. Data augmentation might be utilized to overcome some limitations. The commendable amount of resources and efforts of researchers in the improvement of computer-aided tools have enriched pharmaceutical R&D. Correct implementation and rapid integration of such tools in drug design methodologies can potentially be life-saving. Moreover, understanding the dynamism of the vast diversity inhabiting us is another fascinating aspect worth venturing into.
Article Source: Reference Paper
Learn More:




Aditi is a consulting scientific writing intern at CBIRT, specializing in explaining interdisciplinary and intricate topics. As a student pursuing an Integrated PG in Biotechnology, she is driven by a deep passion for experiencing multidisciplinary research fields. Aditi is particularly fond of the dynamism, potential, and integrative facets of her major. Through her articles, she aspires to decipher and articulate current studies and innovations in the Bioinformatics domain, aiming to captivate the minds and hearts of readers with her insightful perspectives.





{kind=link}