
According to Levinthal’s paradox, each protein may adopt around 10300 distinct structures. We now know 3-D structures for about 98% of the human proteome thanks to DeepMind’s AI system, AlphaFold. However, AlphaFold has limitations, such as its inability to generate coordinates for tiny molecules and ligands that are critical to the protein’s activity. Researchers from the Netherlands Cancer Institute introduce AlphaFill, an algorithm that exploits structural similarity to graft molecules from the experimentally determined structures onto the predicted protein models. The results of more than 12 million transplants performed on more than 995,000 AlphaFold models, along with the validation measurements that went along with them, can be found in the “alphafill.eu” database. This tool is intended to help researchers come up with fresh concepts and create focused experiments.
Requirement for AlphaFill
The process of protein folding is one of structural biology’s “holy grails.” Proteins are fundamental to life, therefore, the consequence of the three-dimensional structure of a protein is of considerable biological significance. Understanding how proteins fold allows a better analysis of various molecular processes and structures.
Artificial intelligence techniques like AlphaFold and RoseTTAfold have made the prediction of protein structures quite accurate, with the exception of the more flexible sections of a protein (loops or disordered areas), which are predicted with lesser accuracy and confidence. These models have been trained using structures that have been experimentally resolved. To fold, myoglobin and hemoglobin, for instance, need heme. The AI models are incapable of predicting the coordinates of the associated smaller molecules or ligands.
By grafting the corresponding cofactors from empirically obtained models, the AlphaFill approach may be utilized to enrich predicted models. The AlphaFill algorithm discussed here has been validated against experimental structures and applied to all AlphaFold models to create a new resource, the AlphaFill databank, which is intended to assist life scientists in easily generating new hypotheses for protein function and formulating relevant research questions. Working of AlphaFill
To find the compounds of interest, sequence homologs for each AlphaFold structure were searched in the PDB-REDO database. PDB-REDO is a database comprising rebuilt and verified entries from the Protein Data Bank. Structures having an identity of more than 25% across an aligned sequence of at least 85 amino acid residues are considered to be hits. As candidates for ‘transplants,’ the most abundant ligands in the PDB and cofactors and their analogs from the CoFactor database are retained.
The hits are structurally aligned on the C-alpha atoms of the BLAST-aligned residues, and the root-mean-square deviation (RMSD) is determined. All backbone atoms within 6 Å of the atoms of each compound, starting with the structural homolog with the closest structural similarity, are considered for “transplantation.” The AlphaFill model is then constructed by transplanting compounds into the AlphaFold model unless the identical compound has already been put within 3.5 Å of the centroid of the compound to be fitted (from a previous homolog). The AlphaFill databank stores all AlphaFill models and metadata. The AlphaFill software is available on GitHub.
Validation
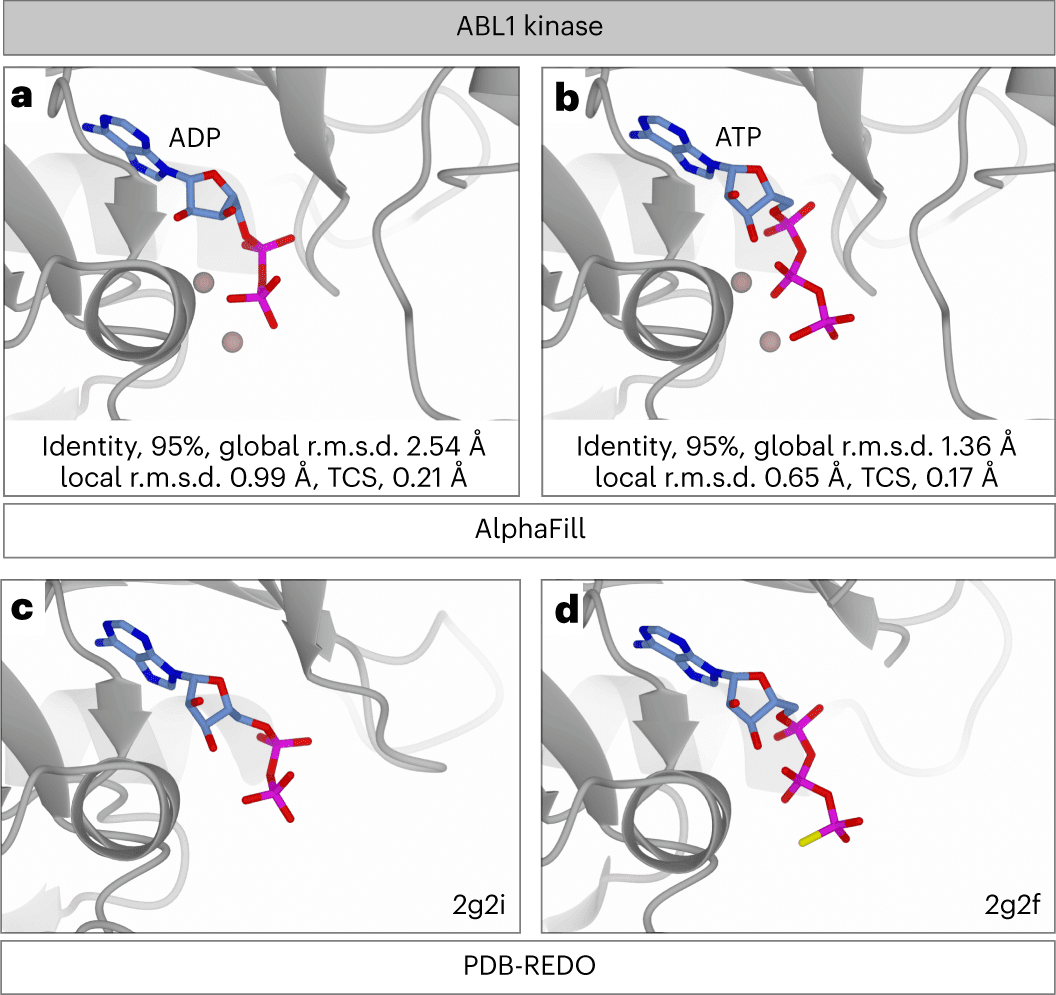
To verify the AlphaFill algorithm, AlphaFill transplants were compared to experimental structures with identical sequences. The local environment validation (LEV) score is defined as the all-atom root-mean-square deviation of any ligand atom and all proteins’ atoms within 6.0 Å of the ligand between the AlphaFill and experimental complexes. This was followed by comparing the LEV scores, which can only be determined when a sequence-identical experimental structure is available, to the local r.m.s.d., which is computed for each transplant according to the aforementioned definition. Local r.m.s.d. decreases with increasing structural similarity, as predicted.
Evaluating potential clashes between ligand and protein atoms is an orthogonal method of validating the transplant’s quality. In order to do this, the transplant clash score (TCS) was formulated as a function of the van der Waals overlaps between a transplanted ligand and its binding site. Since single-atom compounds are overrepresented in the sample and have relatively few collisions, they were omitted from the TCS evaluation to prevent analytical bias. The TCS has a strong correlation with the LEV score. A high TCS may indicate an incompatible binding location, unsatisfactory performance of the AlphaFill algorithm in ligand transplantation, or local inaccuracies in the AlphaFold model.
Determining the quality of models
The validation was then utilized to develop quality indicators that were used to annotate transplants in the AlphaFill database. As the local r.m.s.d. corresponds well with the LEV score, the transplant was annotated by analyzing its distribution as a function of sequence identity. The local r.m.s.d. distribution is rather stable for structures with at least 70% sequence identity. As TCS also correlates well with the LEV score, it was used in a similar fashion.
Web-based UI for AlphaFill
All AlphaFill submissions are accessible for visual inspection on the AlphaFill website. The chosen AlphaFill model is shown using the visualization software Mol, which provides users with complete inspection flexibility. Transplants in the table are colored according to the local r.m.s.d.-based and TCS-based confidence levels. Medium-confidence transplants (marked in yellow) should be treated with care; low-confidence transplants (in red) should be handled with extreme caution. If a single transplant is chosen in the table, the user has the choice to energy-minimize or optimize that specific transplant.
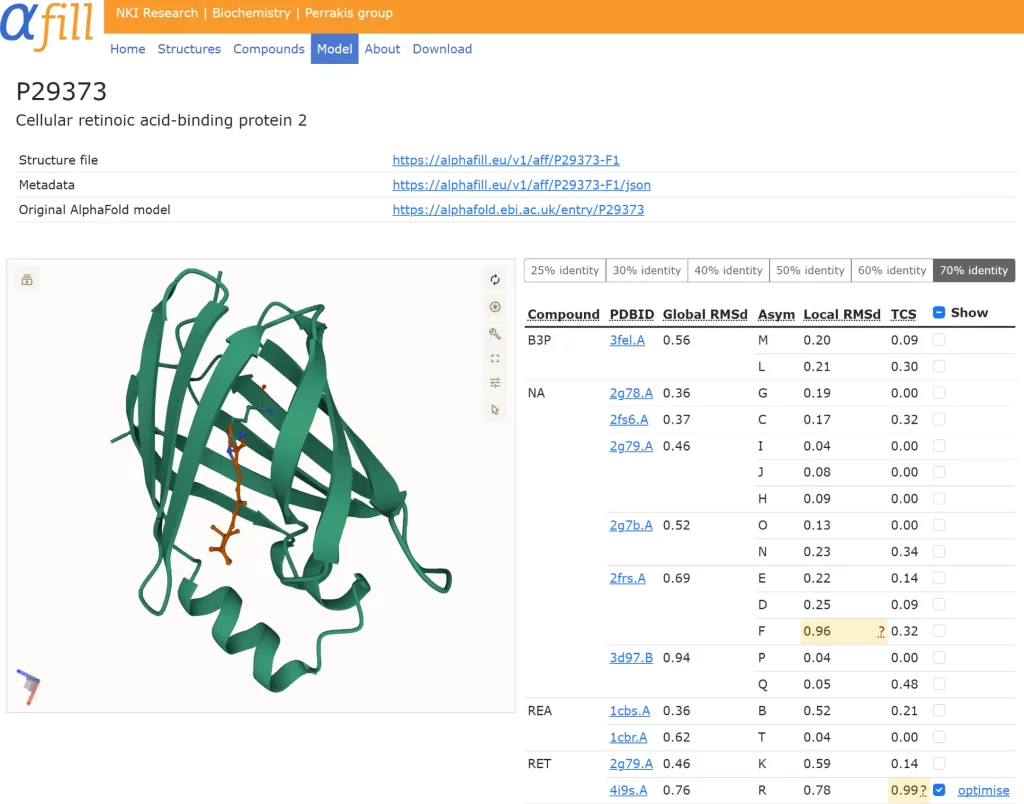
Image Source: https://doi.org/10.1038/s41592-022-01685-y
The case of myoglobin and heme
AlphaFill can transplant compounds from homologous experimental structures that may have been found in another species, as well as to domains for which experimentally proven homologous domains are accessible. Consequently, the databank provides extra capabilities for the annotation of models, which may aid users in making informed decisions on these structures.
Let us consider myoglobin and heme as an example. Myoglobin is a ɑ-helix protein containing heme B as a cofactor that binds molecular oxygen and several other small molecules. The AlphaFold model has a heme-shaped cavity, which is almost equivalent to experimentally determined structures. According to the information in the CoFactor database, several heme analogs are linked back to heme B. Molecular oxygen, and carbon monoxide is added to the AlphaFold myoglobin model as additional components. The latter is attached to two locations: one near the iron atom in the heme and the other on the opposite side of the heme. The second carbon monoxide, positioned in an unexpected location, is inherited from the PDB-REDO entry 1dwt, where it was modeled with a 30% occupancy rate. This occupancy is kept in the AlphaFill model so that users may consider it while assessing the model.
Conclusion
Analyzing the interactions between proteins and cofactors, ligands, and ions facilitates comprehension of both the function and structural integrity of proteins. These compounds are not included in the AlphaFold database, however, PDBe-Knowledge Base provides links for each predicted model to experimental structures. The AlphaFill algorithm does not restrict transplantation to the identical protein but also includes its homologs. AlphaFill structure models are not intended to be exact, precise, or exhaustive representations of the whole repertory of ligands for a particular protein structure. AlphaFill relies on high-quality structural homologs as the main criterion for ligand transfer. AlphaFill could be improved by structure-based algorithms based on deep learning ideas similar to those used in the AlphaFold structure prediction revolution.
Article Source: Reference Paper | AlphaFill: Webserver
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Sejal is a consulting scientific writing intern at CBIRT. She is an undergraduate student of the Department of Biotechnology at the Indian Institute of Technology, Kharagpur. She is an avid reader, and her logical and analytical skills are an asset to any research organization.





{kind=link}