
Researchers from Penn State University present CLIMB (Composite Likelihood eMpirical Bayes), a statistical tool that discovers patterns of condition specificity in a given genomic data. It also provides a generic framework for a variety of investigations, such as aggregating genomic features with comparable condition-specific patterns. CLIMB outperforms the existing options in statistical precision while capturing interpretable and biologically important clusters in the data.
Identifying variations across diverse biological circumstances has been a recurring subject in large-scale genomic data analyses. Many genome-wide association studies (GWAS) have examined the tissue-specificity of gene expression patterns, differential protein binding across cell types, and causative single nucleotide polymorphisms (SNPs) and pleiotropic genetic variants. Two contexts of GWAS served as motivating factors:
1) If a set of subjects has been seen in various settings, one may attempt to classify them based on the patterns of association they demonstrate across biological conditions. When exploring the plasticity of gene expression across many human tissues, for example, a collaborative analysis of this data can be performed to ascertain the groups of genes are collectively up-regulated in specific tissues but down-regulated in others.

Image Source: https://doi.org/10.1038/s41467-022-34360-z
2) Which signs are consistent across investigations? For example, if numerous ChIP-seq datasets are collected under various experimental conditions, one might query whether loci are consistently bound in a certain number of those conditions.
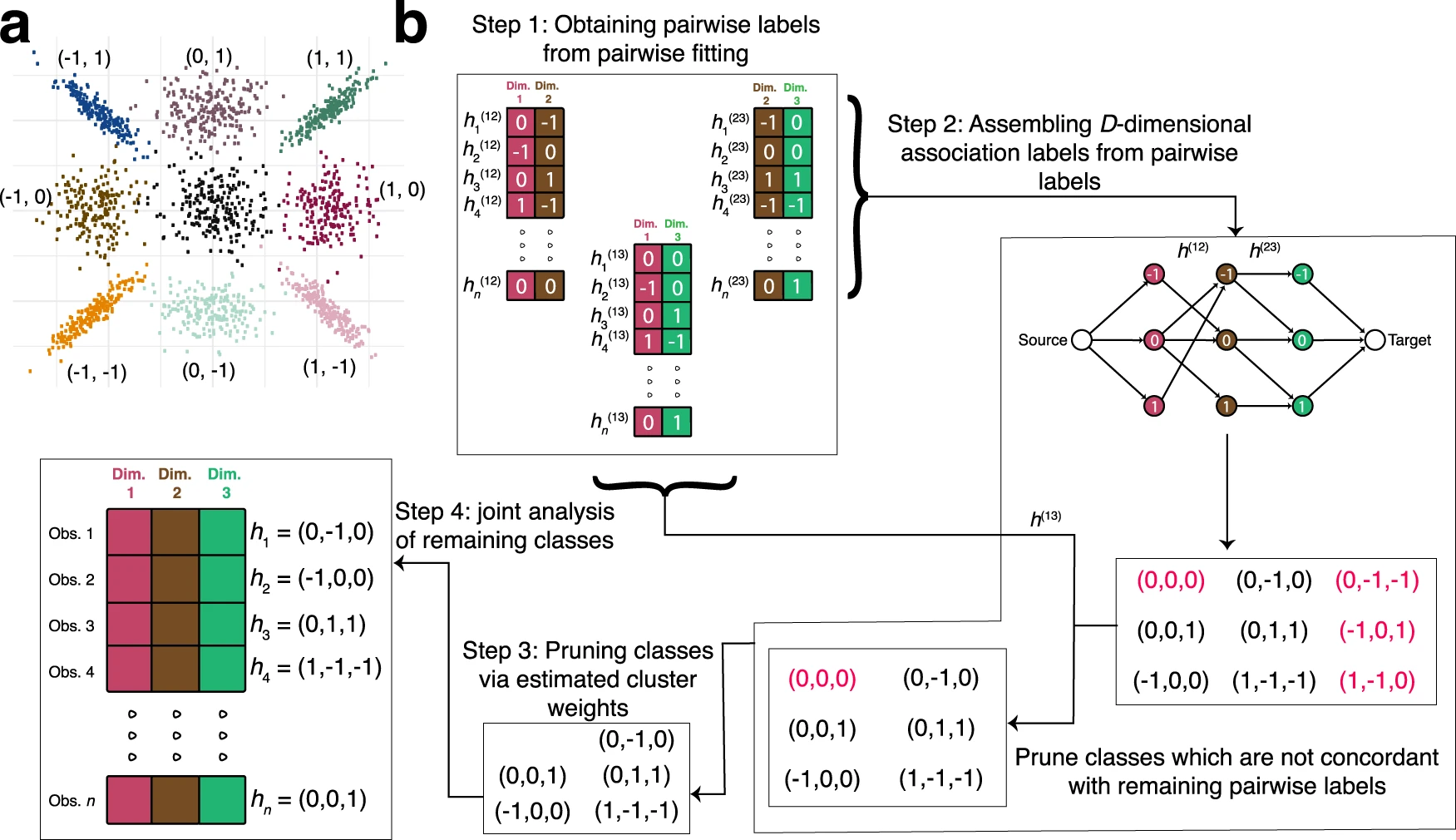
Both motivating contexts are concerned with identifying observations with either null or substantial relationships across a set of conditions. One common method for assessing a set of conditions is K-means or hierarchical clustering. However, this method does not discriminate between an observation that is significant in two circumstances in opposite directions and an observation that demonstrates a consistent direction of association across conditions.
To address downfalls in genomic clustering analysis, the authors Koch et al. introduce CLIMB, a methodology that estimates which latent association vectors are likely to exist in the data. The data used include ChIP-seq, RNA-seq, and DNase-seq data from blood cell lines; the authors show that CLIMB outperforms existing alternatives in terms of statistical power, precision, and model interpretability for studying cell type-specific protein binding and chromatin accessibility, as well as lineage-specific gene expression patterns.
Demonstrating CLIMB’s Value Through Evaluating Multiple Datasets
CLIMB uses CTCF (Transcriptional repressor) binding patterns to discover interrelationships among hematopoietic cell types. CLIMB exposes the predicted links between cell populations and the Bi-clustering heatmaps of all ChIP-seq data for chromosome 11. Similarly, CLIMB was used to conduct lineage-specific differential expression analyses related to cell differentiation and development. CLIMB detects chromatin accessibility patterns in hematopoietic cells related to distinct transcription factor binding signatures.
CLIMB’s capacity to explain distinct patterns of condition-specificity in a mixture with appropriate association vectors estimated from data is a significant advantage. The model is scientifically decipherable thanks to these association vectors. Estimated model parameters can reveal similarities and interrelationships and describe representative association patterns found across experimental settings. Notably, the association vectors serve as the foundation for a novel and effective method of verifying signal consistency across multiple situations or biological studies.
Way Forward
Although the author has concentrated on specific molecular markers, CLIMB can be useful in other applications, such as multi-omics molecular QTLs (Quantitative trait locus) analysis, while CLIMB’s current implementation allows a few hundred conditions for genome-wide studies, future research will look on algorithmically faster implementations, such as variational Bayes fitting for the final Bayesian mixture model, to support a higher number of conditions.
Article Sources: Reference Paper | CLIMB R Package: GitHub
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Shwetha is a consulting scientific content writing intern at CBIRT. She has completed her Master’s in biotechnology at the Indian Institute of Technology, Hyderabad, with nearly two years of research experience in cellular biology and cell signaling. She is passionate about science communication, cancer biology, and everything that strikes her curiosity!





{kind=link}