
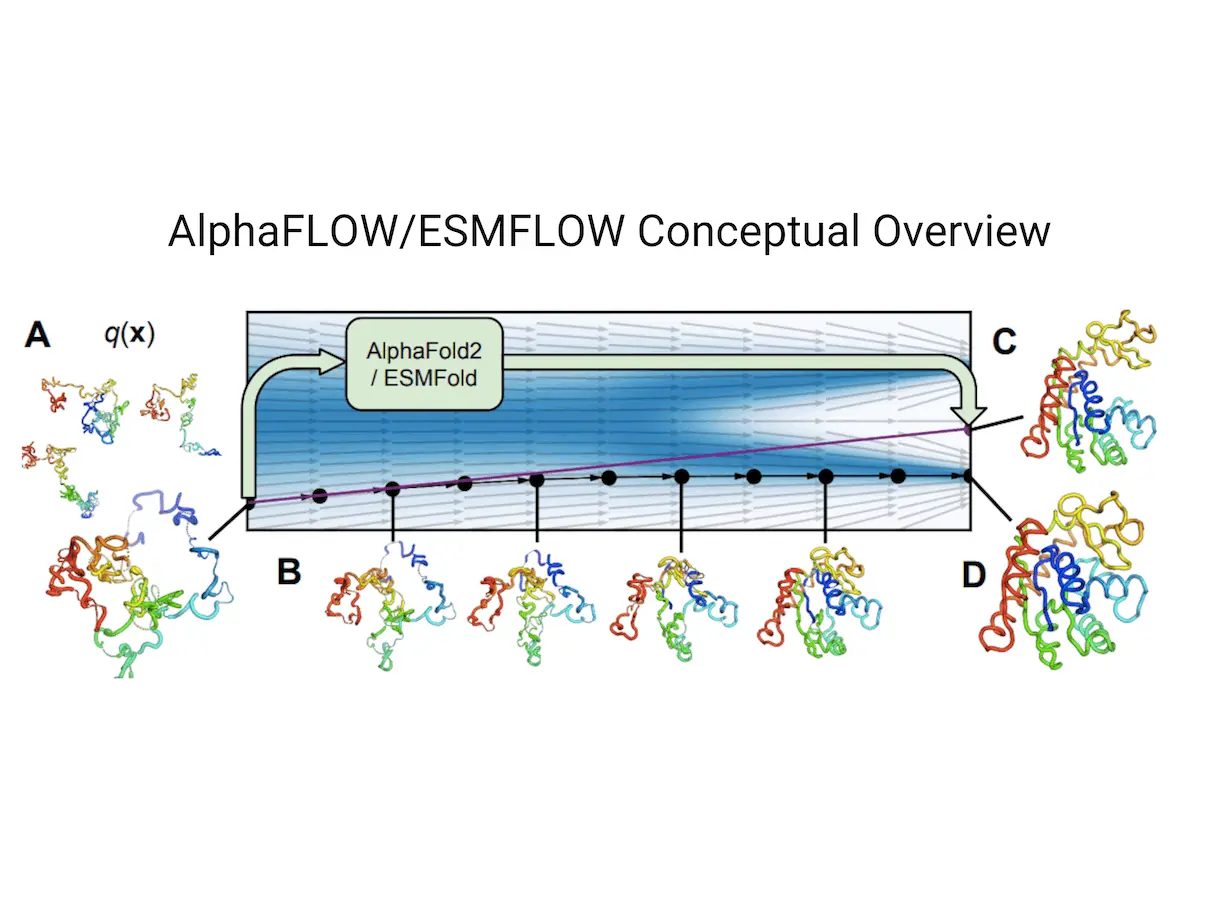
Dynamic structural ensembles play a major role in the biological functions of proteins. For the purpose of learning and sampling the conformational landscapes of proteins, researchers create a flow-based generative modeling technique in this work. Sequence-conditioned generative models of protein structure are produced by optimizing the highly accurate single-state predictor AlphaFold technique within a unique flow matching framework. This approach outperforms AlphaFold with multiple sequence alignment (MSA) subsampling in terms of precision and diversity when trained on the PDB. For proteins that are not yet known, it precisely records positional distributions, structural flexibility, and higher-order ensemble observables. The technique has the potential to serve as a stand-in for costly physics-based simulations because it can diversify static PDB structures with faster wall-clock convergence.
Introduction
In order to perform their biological tasks, proteins frequently take on complex three-dimensional structures as components of structural ensembles with unique states, collective movements, and chaotic fluctuations. The intensity and selectivity of molecular interactions, including those involving transporters, channels, and enzymes, are controlled by these structures. While deep learning techniques such as AlphaFold are excellent for single-state modeling, they are not suitable for taking conformational variation into account. Structural biologists might benefit from an approach that reveals underlying structural ensembles and builds upon single-structure predictors.
Limitations of Existing Machine Learning Approaches
- They cannot be applied to structure predictors based on protein language models (PLMs), such as ESMFold or OmegaFold, which are more and more well-liked because of their quick runtime and user-friendliness because they only work with the MSA.
- The inference-time interventions lack the ability to train on protein ensembles that are not available in the PDB, such as those from molecular dynamics, which are highly expensive to simulate yet have substantial scientific value.
Understanding AlphaFold Meets Flow Matching
This paper presents a principled approach to generate generative models of protein structure by combining flow matching with two regression models, AlphaFold and ESMFold. These models, which were first created as regression models, are now used as sequence-conditioned generative models to overcome difficulties in predicting protein structures. With minimum changes to the architecture and training objective, iterative denoising frameworks such as diffusion and flow matching offer a basic formula for transforming regression models into generative models. This method may be used to train or fine-tune any ensemble and works just as well for PLM-based predictors.
For protein structures, AlphaFold and ESMFold are using flow matching, a method that has been proven successful for pictures. A unique framework has been created to enhance these structures’ training procedures. The framework improves the performance of the approach by defining a scale-invariant noising process robust to missing and cropped residues through the use of polymer-structured prior distribution from harmonic diffusion.
It has been shown that the flow matching versions of ESMFold and AlphaFold, called ESMFLOW and AlphaFLOW, outperform the precision-diversity Pareto frontier of MSA baselines on a test set of conformationally heterogeneous proteins that were recently deposited. These models can be further trained on the ATLAS dataset of molecular dynamics simulations, allowing them to learn from ensembles outside of the PDB. AlphaFLOW significantly outperforms the MSA baselines in the replication of higher-order ensemble observables, conformational flexibility prediction, and distributional modeling of atomic locations when tested on test proteins that differ structurally from the training set. Instead of costly simulations, this method might be employed to get equilibrium ensembles of solved protein structures.
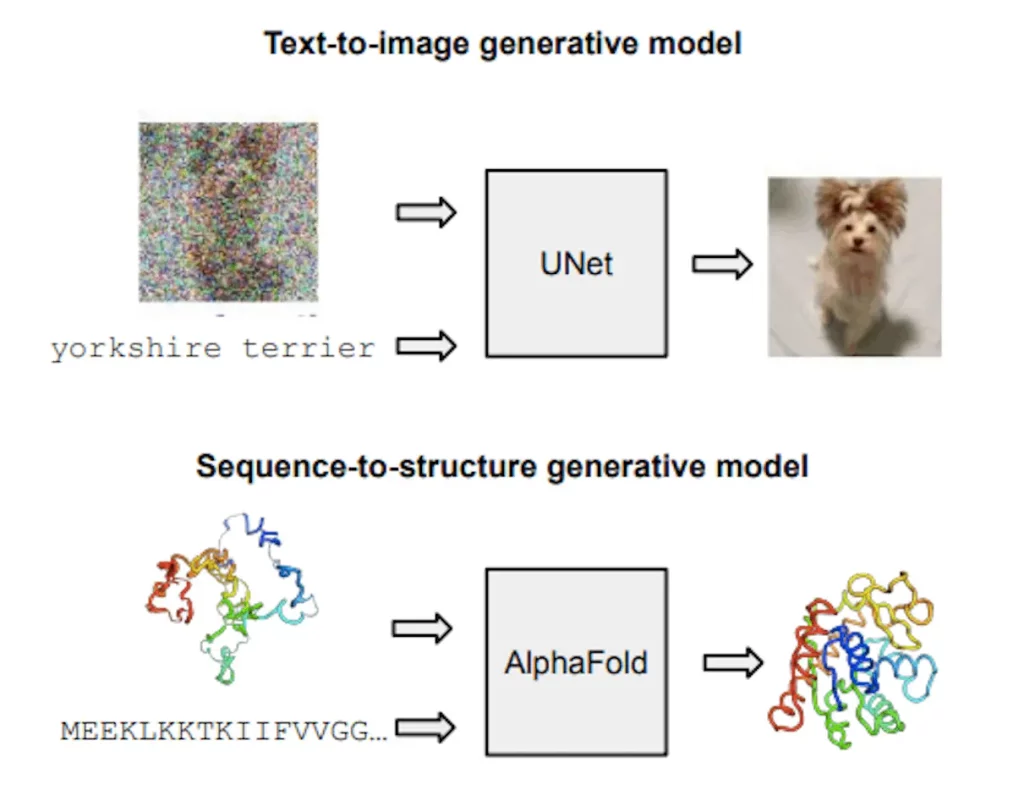
Image Source: https://doi.org/10.48550/arXiv.2402.04845
Looking into Protein Structure Prediction
Using a protein sequence, evolutionarily related sequences, and a template structure, AlphaFold is a contemporary method for predicting protein structures. It predicts all-atom 3D coordinates. This end-to-end technique was created using structures from the Protein Database (PDB) under a regression-like FAPE loss. Subsequent pipeline modifications, such as ESMFold and OmegaFold, avoided template input by replacing the MSA with embeddings from a protein language model (PLM).
Modeling Protein Ensembles: An Overview
The significance of subsampling the MSA input to AlphaFold in order to acquire several functional states was brought to light by Del Alamo’s work in the post-AlphaFold era. This approach has evolved into the accepted paradigm for researching molecular dynamics simulations, variant effects, and kinase conformational states. There have also been suggestions for alternative strategies, including point mutations and clustering. The purpose of the work is to use EigenFold and Distributional Graphormer to train sequence-to-structure generative models of protein ensembles. Ensembles are generated using EigenFold and SE(3) diffusion, and single initial structures are diversified using SENS, a local generative model.
Nevertheless, there have been no compelling validations or comparisons of these models with MSA subsampling techniques on PDB test sets. Learning generative models of Boltzmann distributions as stand-ins for costly molecular dynamics simulations has been the subject of a distinct body of research. Although these models have shown difficulty scaling beyond small molecules and toy systems, they were originally intended to normalize flows. Using MD ensembles as training data, a new generation of generative models with Boltzmann targeting is being built.
What is Flow Matching?
A generative modeling paradigm known as “flow matching” has parallels to and expands upon the notable achievements of diffusion models in the picture and molecular domains. A conditional probability route pt(x | x1), which interpolates between a shared prior distribution and an approximation Dirac distribution, is the fundamental entity in flow matching. The time evolution of pt(x | x1) is generated by a conditional vector field ut(x | x1) and is subsequently trained by a neural network. The learnt vector field is an ODE that evolves to the data distribution pdata(x) from the previous distribution q(x) at convergence. As a special case of diffusion models, flow matching avoids some of the problems that diffusion would often cause.
Conclusion
The new techniques AlphaFLOW and ESMFLOW combine the strengths of AlphaFold and ESMFold to predict accurate and varied PDB structures. These techniques provide a principled training-time approach to represent structural diversity, going beyond changes made to inputs during inference. The outcomes of the experiments show how effective AlphaFold is at forecasting the distributions and characteristics of MD ensembles, both with and without initial experimental structures. It is expected that the generative training paradigm of AlphaFold and ESMFold will find many interesting applications in the field of structural biology, particularly with the growing availability of high-resolution cryo-EM data.
Article source: Reference Paper | Code is available on GitHub
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}