
Millennia of evolution has allowed life to flourish in a million different ways, each suited to the niches it fills. The advent of CRISPR and other genetic engineering methods has allowed science to re-engineer cells to make them even more efficient, robust and fulfill more varied functions. One method for cellular engineering requires the use of genetic interventions that can transform the cell. However, the high cost of determining the optimal mutation necessary for a certain application is an obstacle that hinders the progress of many projects. Scientists from MIT and Harvard have collaborated to reveal a new method that utilizes machine learning, which can reveal the ideal intervention for given parameters, hence saving time and money.
Cellular reprogramming has been the subject of much scrutiny in the field of computational biology. The method, which involved identifying genetic perturbations in order to invoke desired changes in the cell, was proven to be an effective way of engineering cells for human purposes, especially in the fields of immunology and stem cell technology. However, the human genome, which possesses 20,000 genes and more than 1,000 transcription factors, proved not only to be a vast playground for researchers but also the cause of significant expense. As repeated wet lab experimentation was necessary in order to identify the optimal perturbation for the objective, it also proved to be an extremely costly method, which became a major deterrent to scientists who could have otherwise utilized it.
The Introduction of a New Machine Learning Approach
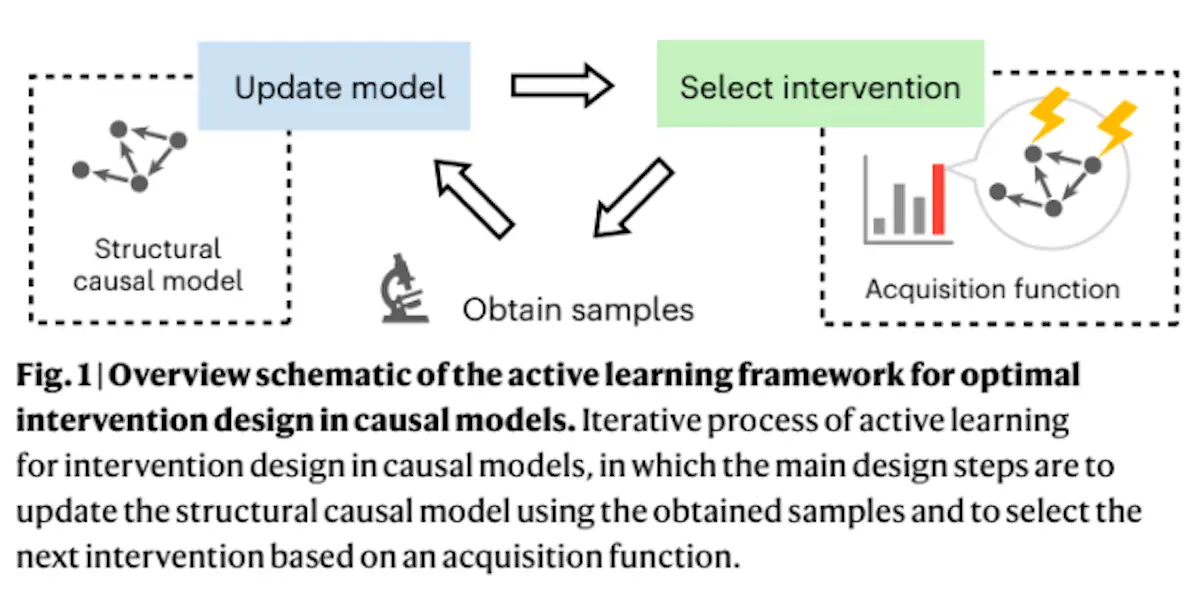
A team of researchers has developed a new strategy for the identification of optimal perturbations to produce a certain outcome – they use a machine learning strategy called active learning that can strategically and sequentially identify new interventions, eventually discovering an optimal intervention using the smallest number of samples possible. Once an intervention is selected, new samples are obtained, which are then uploaded into the model to inform the identification of the next intervention. Here, the model is continuously trained using active learning in an iterative process. It utilizes the relationships that exist between and influence different factors in the system (in this case, gene regulation and expression) such that the best interventions are prioritized. The method, hence, has two ingredients: updating the causal model by utilizing a Bayesian approach and optimizing the effect of the model.
In methods utilizing active learning, acquisition functions are designed that can evaluate potential interventions and select the most optimal one. This is repeatedly used until a single intervention is identified. Most acquisition functions are generic in nature and, as such, can’t be effectively used for problems that are as complex as those found in cellular reprogramming. They also often only take into account the correlation between factors, but not causative relationships or regulatory relationships. This negatively affects their applicability in most fields; in the context of genetic interventions, the inability to take causal relationships into account means the approach will be unable to distinguish between upstream and downstream genes meaningfully, hampering its usefulness.
The novelty of this approach is that it takes into account these causal relationships: the acquisition function is designed so interventions are automatically evaluated on the basis of causal relationships, using available data. Once the experiment has been performed, new data can be utilized to gain a more accurate model, and the process continues, with the search space gradually shrinking. The acquisition function was improved using output weighting – where an optimal intervention is viewed as an extreme event in a complex system.
Experiments were performed on synthetic and biological datasets to determine the validity of the model. In the latter, the method was evaluated on its ability to identify an optimal intervention by which a state change could be induced in melanoma cells – the intervention here was a genetic perturbation that targeted gene expression and reduced their expression to zero (otherwise known as knockout interventions). The model outperformed empirical analogs, hence demonstrably proving that this new method can accurately predict optimal interventions using fewer experiments than current methods. It was more effective at every stage – this means that even with fewer experiments and less data gathered, the results garnered through this approach will still be better than those obtained through conventional methods.
There are, however, some drawbacks of the model: for one, the causal structure was known. In the future, settings may be researched where the causal structure is only partially known or even completely unknown. More agnostic models can also be used, which don’t assume causal sufficiency exists, as the model being discussed did. In the future, artificial intelligence can also be integrated into the model. It is currently being tested in labs to prove its accuracy and veracity further. While the approach was developed for biological applications, it could be utilized in other fields, ranging from fluid mechanics to sales research.
Conclusion
This approach could prove to be a huge leap forward in the field of biotechnology; it has often been noted that the amount of time and money necessary for most research applications makes many research avenues unfeasible for researchers, who often have to work efficiently under budget constraints. However, computational biology has served to bridge the gap between practicality and scientific exploration by providing scientists with the means to efficiently and effectively find solutions and optimize their workflow. Machine learning techniques have ushered in a transformative era in biotechnology, offering powerful tools to unravel complex biological mysteries, expedite drug discovery, and enhance personalized medicine. This approach could help decipher the intricacies of genetic code, aiding in the understanding of diseases and the identification of biomarkers. By analyzing DNA sequencing data and predicting genetic perturbations, it may be possible to unravel the genetic basis of many diseases.
Article source: Reference Paper
Learn More:




Sonal Keni is a consulting scientific writing intern at CBIRT. She is pursuing a BTech in Biotechnology from the Manipal Institute of Technology. Her academic journey has been driven by a profound fascination for the intricate world of biology, and she is particularly drawn to computational biology and oncology. She also enjoys reading and painting in her free time.








{kind=link}