
Scientists from Yangtze University, Jingzhou, China developed a computational tool, “Grit,” to identify the upstream transcription factor binding sites in orthologous genes by the utilization of mixed Student’s t-test statistics.
The transcription factor (TF) regulates the transcription of DNA to messenger RNA or mRNA by binding to upstream sequence motifs. Distinguishing the locations of known motifs in entire genomes is computationally concentrated.
This study presents a computational tool named “Grit” for screening transcription factor binding sites or TFBS by the coordination of transcription factors to their promoter sequences in orthologous genes.
Image Source: Identification of upstream transcription factor binding sites in orthologous genes using mixed Student’s t-test statistics
It utilizes a recently evolved mixed Student’s t-test statistical methodology that recognizes high-scoring binding sites using conservation data among species.
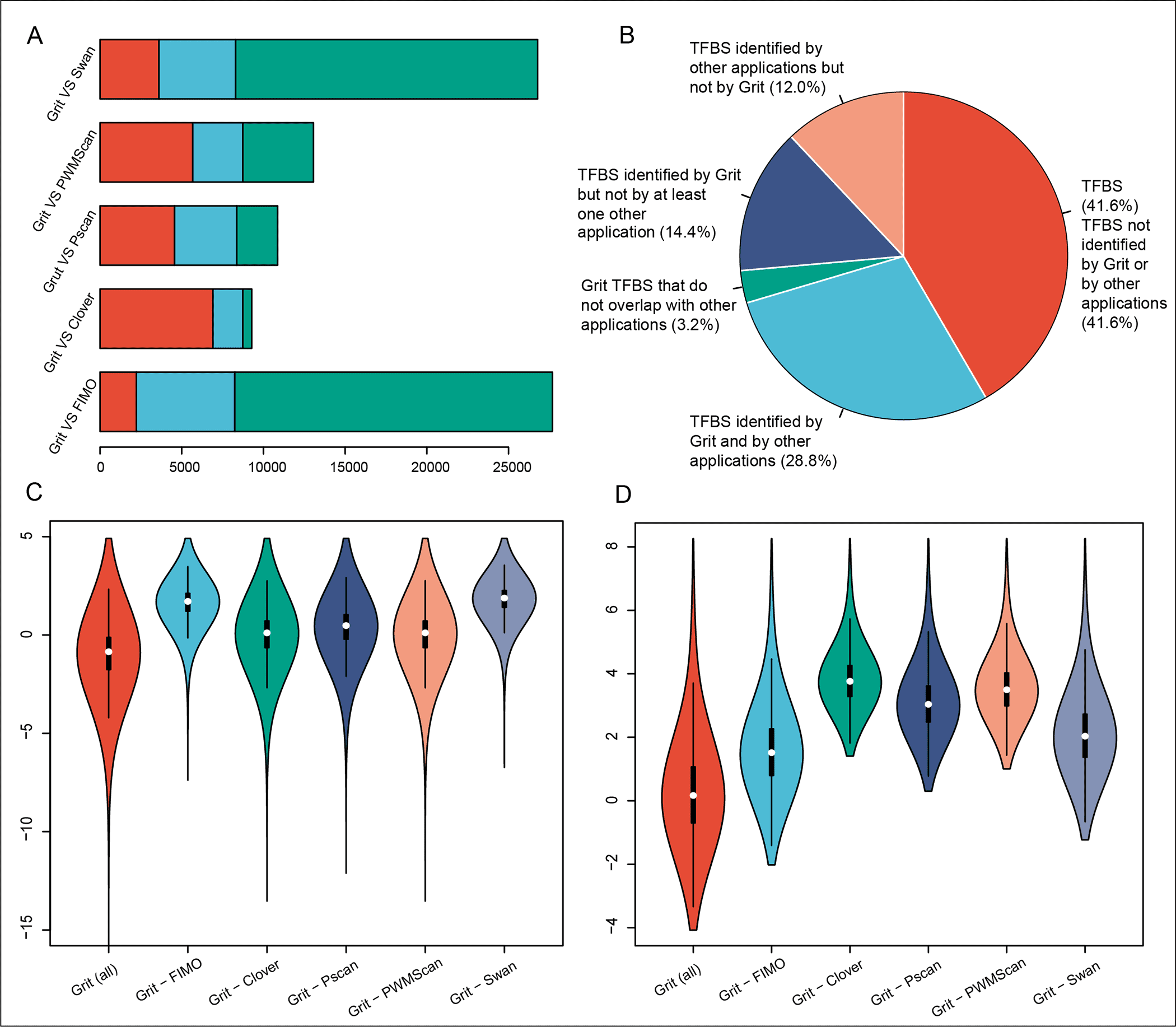
The Grit program performs sequence scanning at a pace of 3.2 Mbp/s on a quad-core Amazon server and has been benchmarked by the deep-rooted ChIP-Seq datasets, putting Grit among the highest level TFBS predictors.
It essentially beats the notable transcription factor motif examining apparatuses, Pscan (4.8%) and FIMO (17.8%), in the analysis of irrefutably factual and well-documented ChIP-Atlas human genome Chip-Seq datasets.
The Utilization of Point Weight Matrices (PWM)
A DNA sequence motif is a short, conserved pattern that can be efficiently coordinated by regulatory proteins, for example, transcription factors (TFs). DNA sequence motifs address functionally significant regions of the genome and are one of the essential units of molecular models that are normally conserved among species.
Finding these motifs in the genome and understanding their capability is crucial in building molecular models for biological processes, for example, human diseases.
Scientists frequently face the task of identifying putative binding sites for transcription factors in entire genomes, called “motif scanning.”
Throughout the course of recent many years, numerous computational pipelines have been portrayed that use point weight matrices (PWM) for tasks like these.
The Functions of Available Motif Scanning Tools
The study describes the functionality of various currently available motif scanning tools in detail. Some of them are explained below:
- MAST: It looks through DNA motifs against a data set made out of short sequences and assigns a score to each target sequence expecting that each motif occurs once.
- MCAST: It utilizes a hidden Markov model (HMM) to scan DNA sequences for regions involving at least one of the given motifs, though SWAN makes use of a log-likelihood ratio or LLR scoring framework built via training a two-state HMM on the background sequences.
- FIMO: It computes an LLR score for each situation in a DNA sequence motif and converts this score to a q-value utilizing dynamic programming techniques.
- TRAP introduces a physical binding model which predicts the relative binding affinity of a transcription factor for a given sequence.
- PWMScan examines sequence motifs utilizing Bowtie or “matrix_scan,” which utilizes a conventional search algorithm.
- The Python-based program Motif scraper searches motifs determined as a text string utilizing IUPAC degenerate bases.
- A few other tools, for example, Toucan, OTFBS, and CREME include all matches in the target and control sequences and apply binomial statistics for over-portrayal.
- Different software, for example, Clover, PAP, oPOSSUM, Pscan, and TFM_Explorer examine sequence sets from co-regulated or co-expressed genes with transcription factor motifs and survey motifs that are fundamentally over or under-addressed to distinguish common regulators of the sequence sets.
- WeederH was intended for finding conserved TFBS and distal regulatory modules in sequences from orthologous genes.
- MatrixREDUCE predicts useful transcription factor binding through alignment-free and proclivity-based examinations of orthologous promoter sequences in closely related species.
Development of the “Grit” Motif Scanning Algorithm
To beat the inadequacies of current accessible tools as described in the study, a novel motif scanning algorithm, “Grit” was fostered that distinguishes genome-wide upstream transcription factor binding site from a known set of PWMs for promoters of orthologous genes without the need for sequence alignment.
The given study resolved the subject of tracking down critical relationships among transcription factors and orthologous gene sets by presenting another computational structure that utilizes mixed Student’s t-test insights and embraces better approaches for developing promoter sequence sets of orthologous genes.
Its application to the human genome has yielded productive outcomes, showing its desirability as a motif scanning tool.
The Endpoint
Finding transcription factor binding sites in the genome and distinguishing their capability and function is basic in the comprehensive understanding of different biological processes.
Improvement of the exhibition of the prediction tools is significant in light of the fact that exact and precise transcription factor binding site prediction can save cost and time for wet-lab tests.
Additionally, genome-wide TF-binding site prediction can give new insights and intensive knowledge for transcriptome regulation from a systems biology point of view.
The given study fostered another TF-binding site prediction tool in light of the mixed Student’s t-test statistical methodology.
The tool is among the highest level transcription factor binding site predictors, thus, it can help the scientists in TF-restricting site identification and interpreting transcriptional regulation mechanisms of genes.
Grit is a decent option in contrast to currently accessible motif scanning and is openly accessible under a scholarly free permit at:
Future directions will be to develop algorithms like gene set enrichment analysis for the analysis of transcriptome data.
Article Source: Huang T, Xiao H, Tian Q, He Z, Yuan C, et al. (2022) Identification of upstream transcription factor binding sites in orthologous genes using mixed Student’s t-test statistics. PLOS Computational Biology 18(6): e1009773. https://doi.org/10.1371/journal.pcbi.1009773
Learn More About Bioinformatics:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}