
FAIR bioinformatics research relies heavily on standardized analysis pipelines. Point-and-click pipeline programs like Galaxy have gradually given way to command-line programs like Nextflow and Snakemake during the past ten years. These frameworks promote faster development, collaboration, and sharing of analysis steps across research communities. This paves the way for smoother data analysis, especially for large consortia working on complex projects.
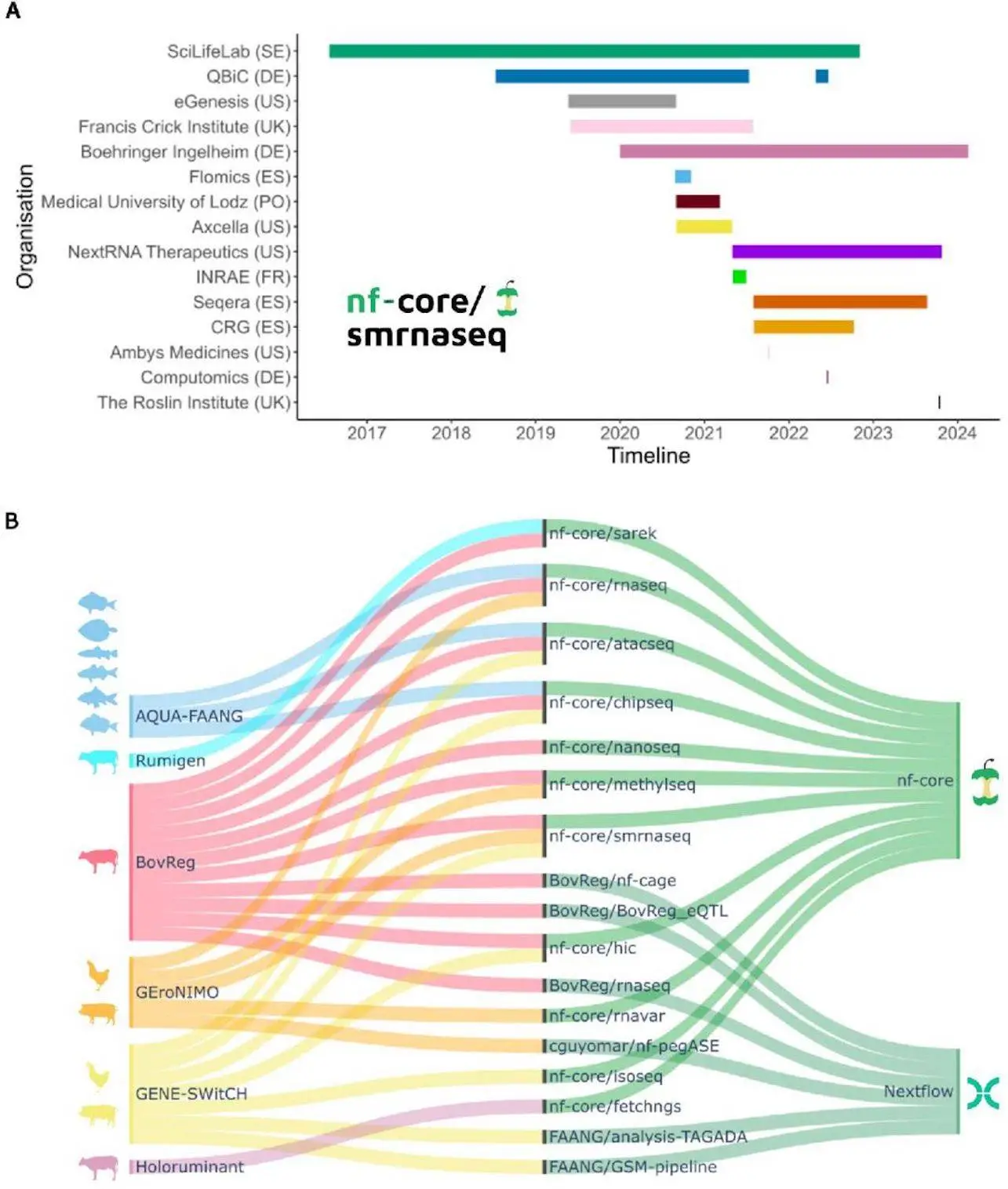
Researchers elucidate how embracing nf-core standards facilitates enhanced interoperability, expedited development cycles, and cooperative efforts among the nf-core community’s over 8,000 members. The Nextflow Domain-Specific Language 2 (DSL2), which was recently developed, allows pipeline components to be shared between European research initiatives. By integrating modules and sub-workflows into any Nextflow pipeline, the nf-core community has made it possible to gradually switch to nf-core best practices. Large consortia were inspired to harmonize data analysis techniques by a case study of nf-core adoption by six European research consortia under the EuroFAANG umbrella, with an emphasis on farmed animal genomics.
Robust and scalable data analysis is required due to the tremendous rise in data generated by advances in molecular biology technologies. The current preferred option for high-throughput data analysis pipelines is workflow management systems or WfMSs. The tools Snakemake and Nextflow, which combine the expressiveness of Bash with extra capabilities to facilitate reproducibility, traceability, parallelization, and portability across different infrastructures, are intended for bioinformaticians with programming experience. More than 2.5 times as many citations were found in popular WfMSs in 2023 than there were in 2018, with Snakemake and Nextflow showing a clear trend of popularity over the previous six years. About a third of reported WfMS usage in 2023 came from Nextflow, the resource with the largest usage growth.
Two potent workflow technologies that make it possible to bundle methods into pipelines are Snakemake and Nextflow. They don’t have any standards for this procedure, though. They have created pipeline registries, including the Snakemake Workflow Catalogue and Nextflow, to solve this issue. These registries offer best-practice standards and enforce implementation guidelines. Nextflow adoption has been greatly aided by the nf-core community, whose curated pipelines have been able to execute 83% of the time without crashing.
The Role of Nextflow and nf-core in Bioinformatics
The process of implementing a bioinformatics pipeline standard at the level of a consortium comprising more than 80 universities and businesses is described in this study by academics, with a particular emphasis on computational analysis. Here, researchers make the case for “soft standards,” or a framework that allows for a range of implementation states, from the most ideal to the present. Because nf-core is built on Nextflow, which can encapsulate any existing code regardless of its implementation language, researchers found it to be a good trade-off. For a Nextflow pipeline to function, strict adherence to nf-core best practices is not required, allowing for gradual evolution towards nf-core standards. Secondary benefits from community participation offset the additional development effort: nf-core pipelines are more transparent and have more accessible documentation. They gain from validation, issue fixes, community support, and extension. These elements are particularly crucial when thinking about sustainability over the long run.
Expansion of the nf-core Pipeline Community and Broadened nf-core Outreach
The nf-core community was founded in 2018 and consists of a selection of pipelines that are applied in accordance with best practices. They have about 100 pipelines that cover a wide range of data types, including microscopy, protein structure prediction, mass spectrometry, high-throughput sequencing, analysis of satellite imagery, and economics. 8,000 Slack users, 100 universities, and over 2,000 GitHub contributors all support these pipelines. Teams from different locations can contribute based on their capabilities and needs thanks to the effective cooperation approach.
In order to promote sustainable growth, Nf-core plans activities and programs for the community. With 100 webinars, 13 virtual hackathon events, and a mentorship program, the organization has drawn participants from all over the world. Additionally, it offers free community training films that, since October 2022, have received over 35,000 views on YouTube. The organization works to ensure that everyone can attend these events, irrespective of language, location, time zone, or ability to travel.
The Nextflow Workflow Management System’s Evolution
Notable advancements in Nextflow pipelines have been made recently, such as the addition of the DSL2 syntax, which makes it possible to divide intricate workflows into reusable parts. The encapsulation and reuse of analytical stages shared across pipelines have been greatly enhanced by this. Further benefits for Nextflow include expanded support for numerous HPC schedulers and software container engines, as well as enhanced support for cloud-based computing platforms and on-demand container provisioning through the Wave service. The uptake of community-driven projects has accelerated, such as nf-test, a testing framework for Nextflow components and pipelines. Its fundamental functionality has been further improved by Nextflow plugins like nf-validation, nfco2footprint, and nf-prov, which has led to its widespread acceptance in disciplines including economics, astronomy, and earth science.
Essentiality of Interoperability and Standards
Technical differences that may arise from the use of different parameterization and analysis tools are prevented by the effective standardization technique offered by nf-core. Results can be compared thanks to this standardization. To make use of the epigenetic states and genomic regulatory elements that have been conserved across evolution in six different species of farmed fish, AQUA-FAANG, for example, used nf-core pipelines to analyze the tissue panels that matched, as well as various biological circumstances. Given that efforts like the farm animal GTEx are anticipated to continue incorporating deeper sequencing and a greater number of species over time, data integration across studies is essential. Long-term sustainability, therefore, depends on interoperability, which is attained by applying a common analytical framework, and this extends well beyond the objectives and lifespan of initiatives such as EuroFAANG.
The Primary Obstacles to an Integrated Structure for Bioinformatics Analysis
One of the most difficult objectives in bioinformatics is currently establishing universal standards. Adopting new standards may include retraining personnel in addition to re-implementing and re-validating methodologies that are currently in use because research organizations frequently have established protocols. It can be advantageous for a chosen leader to lead the standards adoption process. Following the AQUA-FAANG consortium’s decision to employ nf-core, the University of Edinburgh and the Norwegian University of Life Sciences assumed duty for the framework’s deployment and for instructing the other AQUA-FAANG units on how to utilize nf-core pipelines. Whichever framework is selected, a gradual transition has a higher chance of preventing group fragmentation. For this reason, the transition roadmap is just as crucial as the destination.
Conclusion
The nf-core community is expanding quickly, which highlights the value of having rigorous best practice guidelines along with user and developer tooling. The infrastructure of modules and sub-workflows makes it easier for developers to collaborate within nf-core as well as with external pipelines, which improves code quality and extends maintenance life. Collaboration, enhanced interoperability, standardization, and speedier development are made possible by nf-core standards and community support. These standards continue to facilitate effective collaboration and the production of more dependable research among various remote communities as their usage expands beyond traditional bioinformatics into other scientific domains. The successful collaboration between EuroFAANG and nf-core has produced reciprocal benefits and genuinely FAIR science. In an effort to promote more partnerships like this, nf-core has launched a new program called “special interest groups,” where researchers with similar interests can work together to standardize the use of nf-core pipelines. Researchers hope that these groups will operate in tandem with current groups working on particular pipelines, promoting better global scientific cooperation and standards.
Article Source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}