
Making synthetic protein sequences from protein structures is a powerful use of protein engineering technology. It can be challenging to create designed proteins, though, because poor codon sequences can result in low expression rates in organisms like yeast. Scientists from CSAIL-MIT and Harvard Medical School present CodonMPNN, a system that produces a codon sequence conditional on an organism’s label and the structure of a protein backbone. It has been demonstrated that when DNA sequences are near codon optimality, the machine learning method CodonMPNN will produce greater expression yields for codon sequences. Test results indicate that CodonMPNN maintains its prior inverse folding performance and recovers wildtype codons more often than baselines, raising the possibility of producing high-fitness sequences.
Introduction
Protein engineering faces a significant barrier in the expression of engineered RNA in host systems like yeast due to the suboptimality of the DNA/codon sequence used to express the protein. Multiple codon sequences can encode the same protein sequence, but each can interact differently with its environment, leading to different behaviors and expression levels. This suboptimality is a bottleneck in the protein engineering approach of generating protein sequences conditioned on a protein backbone structure using tools like ProteinMPNN. However, these optimization methods are imperfect and often fail for less well-studied hosts, making it difficult to validate the proteins obtained from these tools.
Moreover, a chance for optimization toward greater expression yields is lost in the procedure that follows:
- Obtain the protein structure with the predicted desired function.
- Create an amino acid sequence that folds into the structure.
- Obtain the best possible codon sequence for the amino acid sequence.
It is impossible to modify the amino acid sequence in the previous approach to increase the expression yields of the codon sequence that codes for the structure.
Related Research
The term “inverse folding,” which has a distinct interpretation within the field of biology, has been modified to signify the process of producing a protein sequence that is dependent on the structure of a protein. Many generative models with various factorizations of the sequence distribution and architectures have been presented for this job and have shown promising results in protein engineering. The information regarding the host system in which the modified protein would be produced is not utilized by these existing methods.
CodonMPNN: An Overview
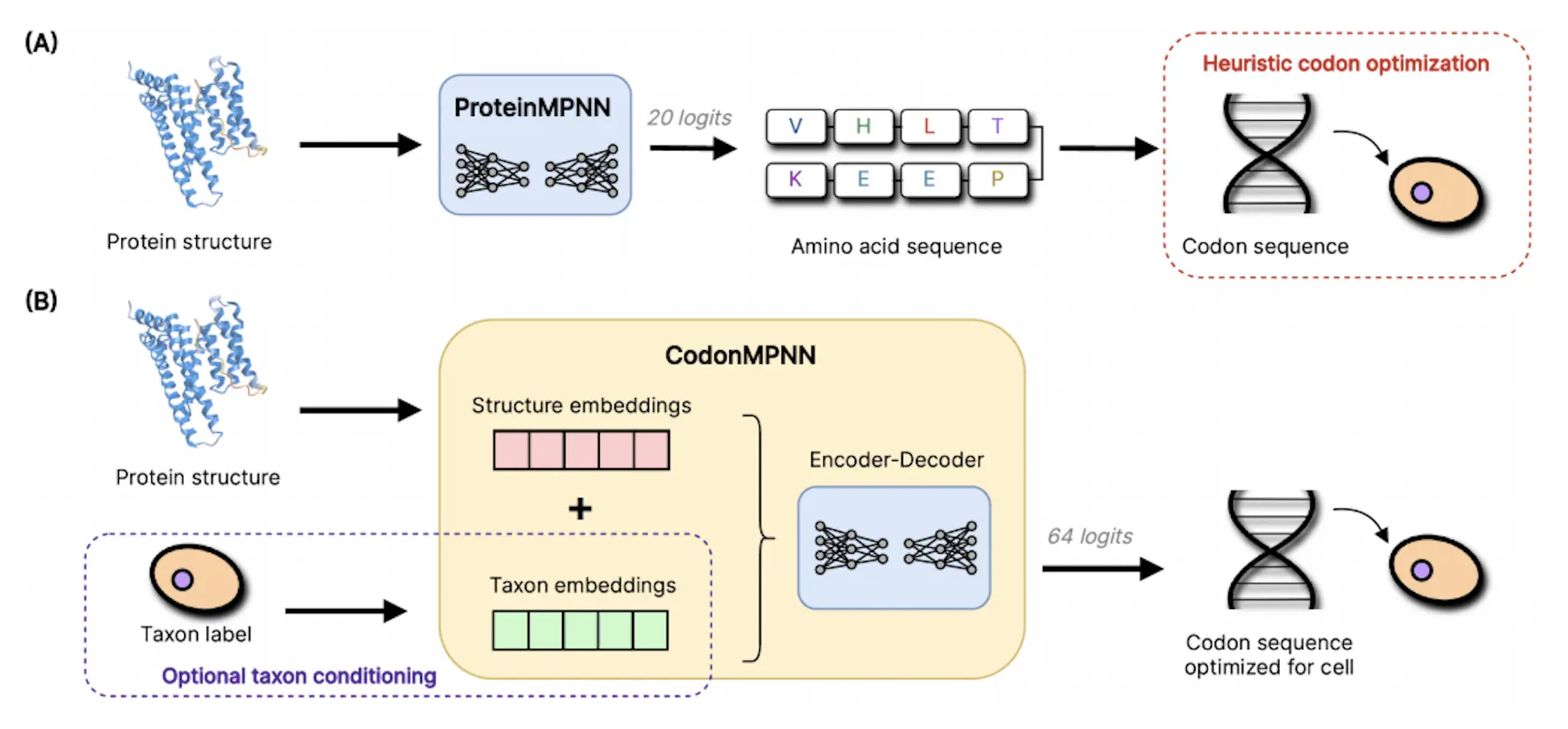
CodonMPNN is a new technique that generates codon sequences from a protein backbone structure and an organism label specific to the host system. This technique generates high-expression codon sequences once trained on natural protein codon sequences. Using the AlphaFold database as its training set, CodonMPNN exhibits comparable recovery rates and designability evaluation metrics to ProteinMPNN. When codon sequences are converted to amino acids and the most frequent codon is selected, the resulting sequences have a lower codon recovery rate. Furthermore, for high-fitness codon sequences, CodonMPNN has a better likelihood.
CodonMPNN is based on the same architecture as ProteinMPNN, but it can predict 64 codons instead of 20 different types of residues. It also has the ability to condition the host system where the DNA is intended to be expressed. As a result, CodonMPNN is an autoregressive model of any order over codon sequences conditioned on protein structures.
Architecture of CodonMPNN
Researchers employ the encoder-decoder architecture of ProteinMPNN for CodonMPNN, in which the encoder predicts using the partial codon sequence and structure embeddings generated by the encoder given the protein structure x as input. Three message-passing neural network layers make up the encoder and decoder, both placed on a graph with residues acting as nodes. Every node has edges with the 48 nodes that are closest to it in terms of the Euclidean distance between alpha carbons. The edge features derived from all pairwise distances between the backbone atoms of connected residues are the sole information derived from the protein structure next to the graph connectivity. As a result, the anticipated probability holds true for every Euclidean transformation of x. By hiding messages from residues with a higher index in the permutation σ than the residue that receives the messages, researchers can execute the autoregressive prediction of the decoder.
Future Work
The objective of many codon optimization tasks is to find the most highly expressed codon sequence for an amino acid sequence for a particular host system, even in the absence of protein structure. Although CodonMPNN can serve as a convenient stand-in for inverse folding models like ProteinMPNN, it is not effective in handling this particular task. Researchers are working to refine a protein language model to create codon sequences that are conditioned on amino acid sequences and their taxon labels.
Conclusion
ProteinMPNN can be swapped out for CodonMPNN, which allows for structure-based protein engineering using inverse folding techniques. Rather than randomly selecting the most frequent codon for each amino acid, it constructs codon sequences directly, bringing us closer to codon optimization. An aspect of CodonMPNN that was absent from earlier inverse folding techniques is the ability to induce the host system to express the produced codon sequence. Tested in the lab, CodonMPNN maintains ProteinMPNN’s functionality while giving more weight to codon sequences that are heavily expressed.
Article Source: Reference Paper | Code is available on GitHub.
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}