
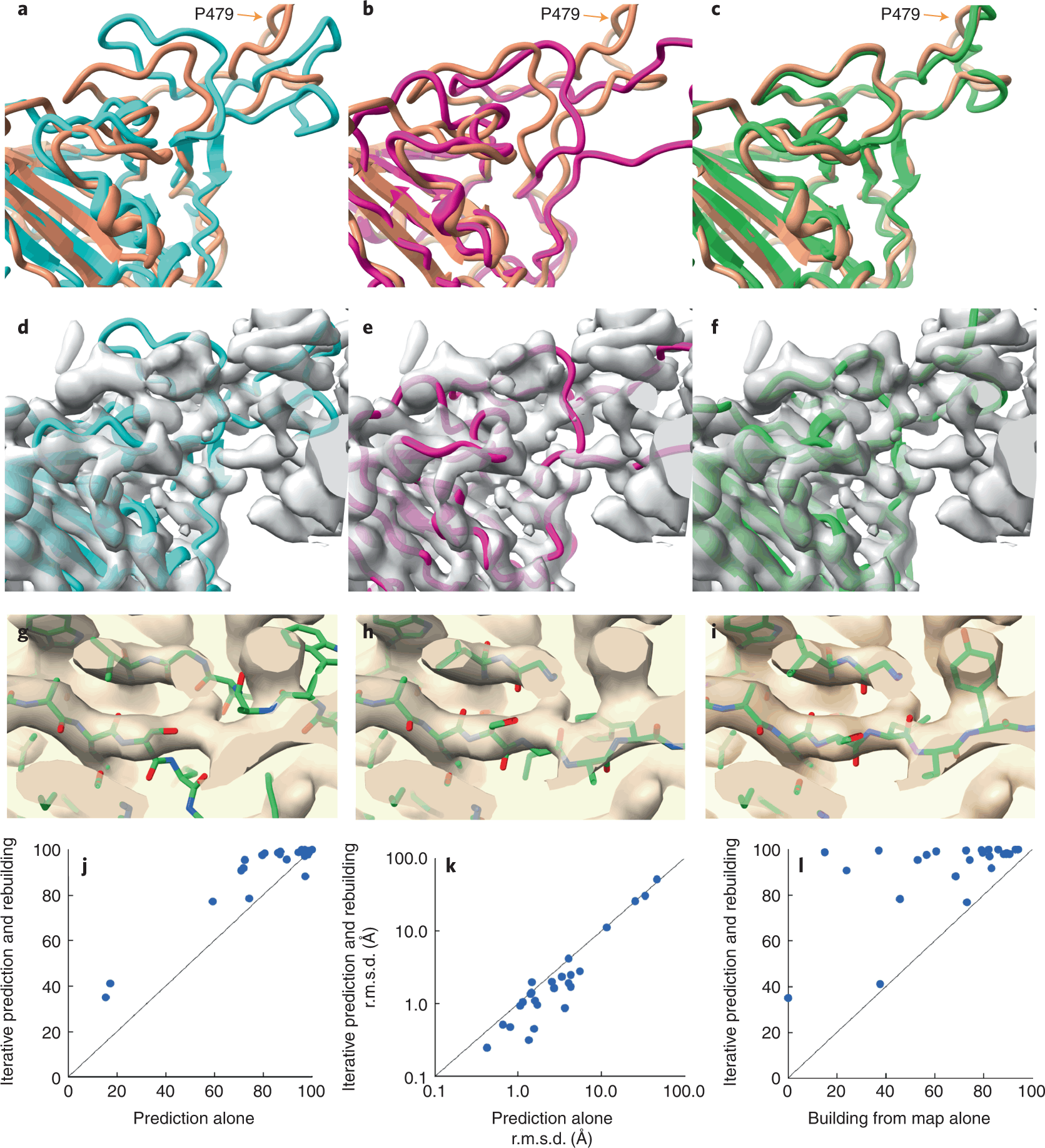
A team of researchers led by Thomas Terwilliger from the New Mexico Consortium developed a procedure where the AlphaFold structure prediction models can be automatically rebuilt based on density maps. These models might, therefore, be used as templates for generating new AlphaFold models in the future.
Machine learning algorithms are widely implicated in the prediction of protein structure. Protein structure prediction is a complex problem since various modifications and possibilities arise due to four levels of protein formation, i.e., primary, secondary, tertiary, and quarternary structure.
AlphaFold and RoseTTAFold can predict the final protein structure using the protein’s primary amino acid sequence. But the limitations of such a machine learning model are that they do not yield accurate predictions for some disordered segments present in a protein, and these segments might adopt an alternative conformation. And although not explicitly mentioned, these algorithms use structures from the protein data bank for training, causing bias in the structure predictions. A computational model like AlphaFold can assess the accuracy of their predictions, where for each residue in a protein molecule, a confidence measure of pIDDT is given.
With this information in mind, the current study puts forth the hypothesis that providing the model with experimental data, such as density maps and crystallographic structures, might result in adjustments in the conformation and improved accuracy of the predicted model. This leads to the proposal of a novel iterative procedure wherein the information in the electron density maps can enhance structure prediction with high-accuracy modeling.
Findings
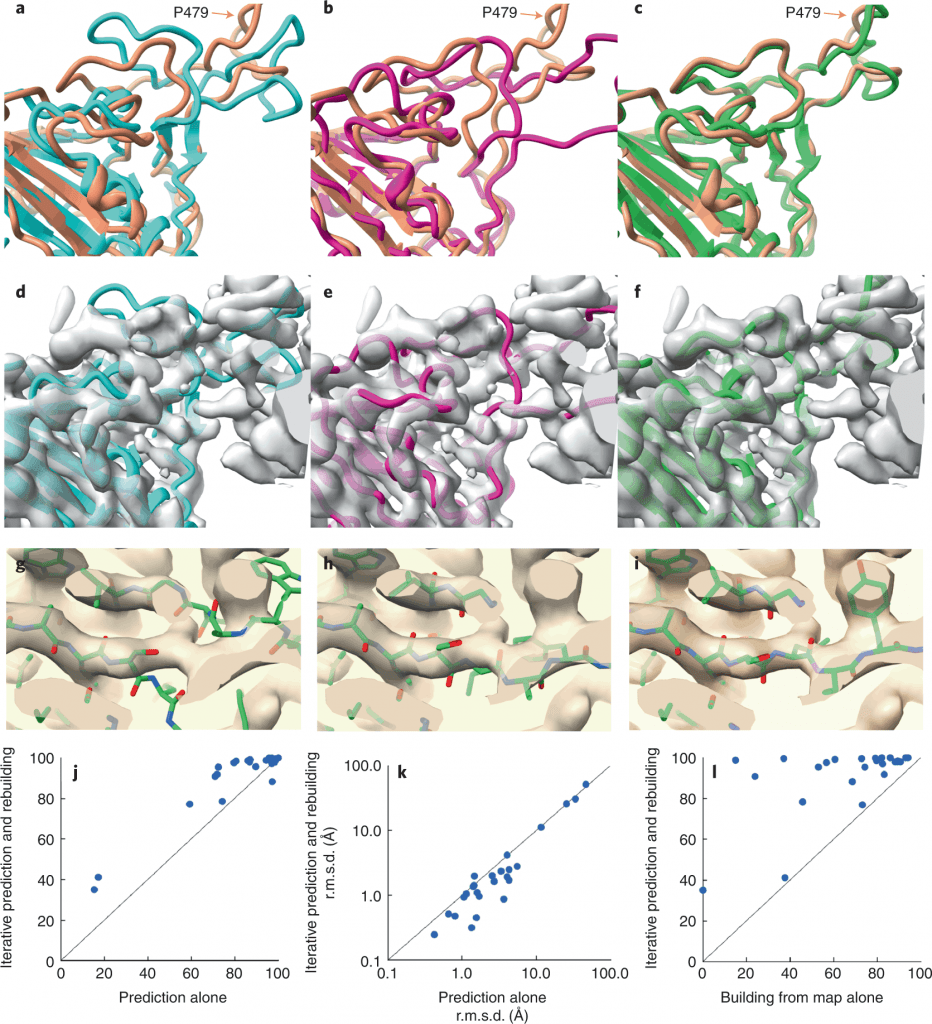
Image Source – https://doi.org/10.1038/s41592-022-01645-6
Researchers retrieved the SARS-CoV-2 spike protein receptor binding domain (PDB ID – 7MLZ) to predict and rebuild the protein model using AlphaFold and Electron Density Map.
As seen in the figure, neither the AlphaFold predictions alone nor the structure built from the electron density map alone gave structures similar to the one deposited in PDB. There were regions where the predicted model did not superimpose the deposited model, bringing about poor accuracy. But when the structures were modeled using an iterative structure prediction procedure using AlphaFold followed by rebuilding using superimposed electron density maps, the predicted structure was highly similar to the deposited structure.
Next, the scientists set out to find if AlphaFold can determine the structure of a protein whose model is not deposited in PDB yet, on which the AplhaFold was trained. Therefore, a bacterial flagellar basal body domain was considered with structures retrieved from PDB at only 9% structural similarity. A similar iterative pipeline of the now-untrained AlphaFold, rebuilding using electron density maps and multiple sequence alignment, was laid. The model predicted was comparable to the closely resembling (9% similarity) structure present in PDB. So this assures the fact that this approach can help unravel proteins whose structures are not present or trained on by PDB and AlphaFold.
Final Remarks
Terwilliger et al. have developed a synergistic model utilizing AlphaFold and Density Maps to help researchers predict unknown and untrained protein structures.
In the first test case, the polypeptide chains were predicted with AphaFold, then only with a density map, and the best fit was developed by combining the predictions with the density map. At the same time, the presence of previously solved protein templates enhanced AlphaFolds’ predictive abilities, but no templates were used in the present study. The authors suggest that including templates representing a part or all of the structure to be modeled could prove to be highly useful. The low-resolution range in which the method is useful may be further expanded as model-building tools improve. Additionally, the low-resolution range may be increased with currently available tools by merely refining and avoiding attempting to recreate structural elements that do not match the density map.
Article Source: Reference Paper | Code availability: Phenix version of the AlphaFold2 Colab | Phenix Code
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Shwetha is a consulting scientific content writing intern at CBIRT. She has completed her Master’s in biotechnology at the Indian Institute of Technology, Hyderabad, with nearly two years of research experience in cellular biology and cell signaling. She is passionate about science communication, cancer biology, and everything that strikes her curiosity!





{kind=link}