
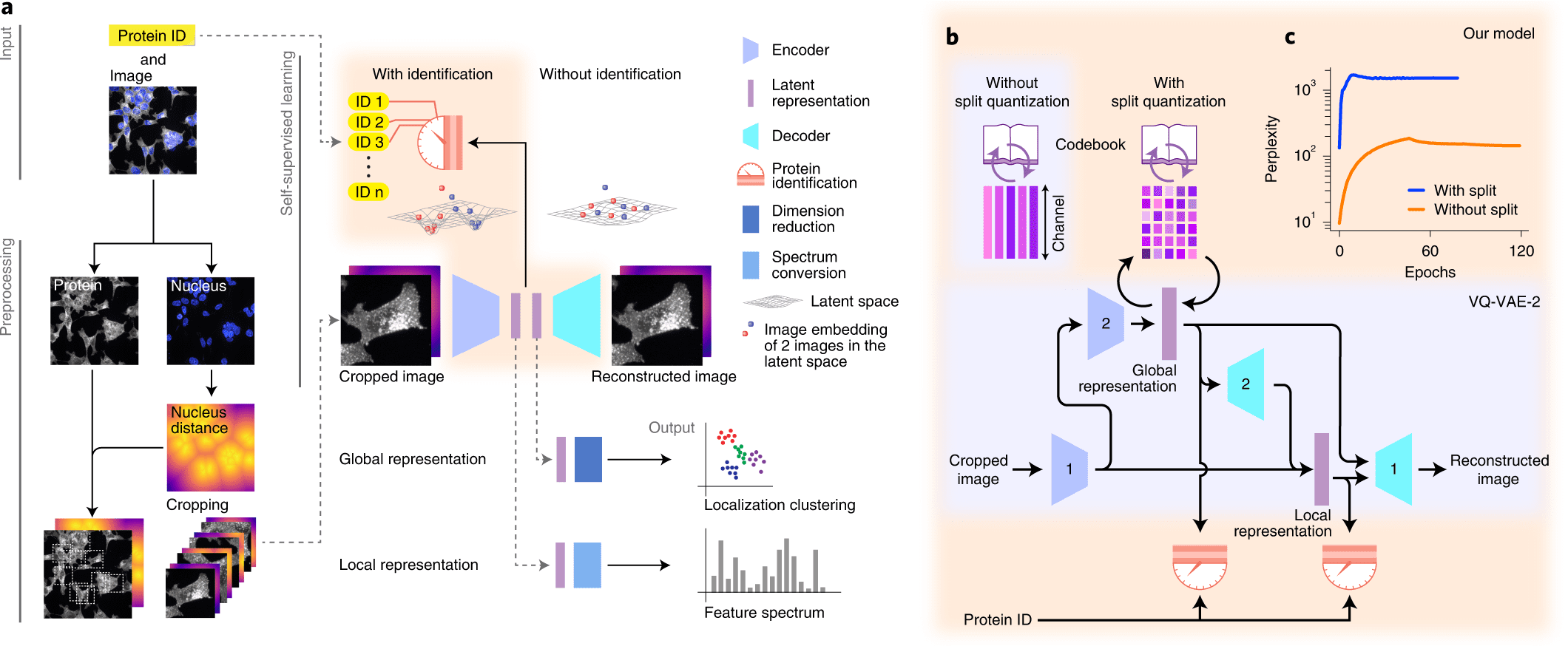
Understanding cellular architecture requires understanding the diversity and complexity of protein localization. The present study aims to introduce cytoself, a deep-learning approach that allows self-supervised profiling and clustering of protein localization.
A self-supervised training scheme is leveraged by Cytoself, which requires no preexisting knowledge or categories. Using images of 1,311 endogenously labeled proteins from the OpenCell database, cytoself reveals a highly resolved protein subcellular localization atlas that summarizes the major scales of cell organization, from nuclear and cytoplasmic classes to subtle localization signatures of individual proteins.
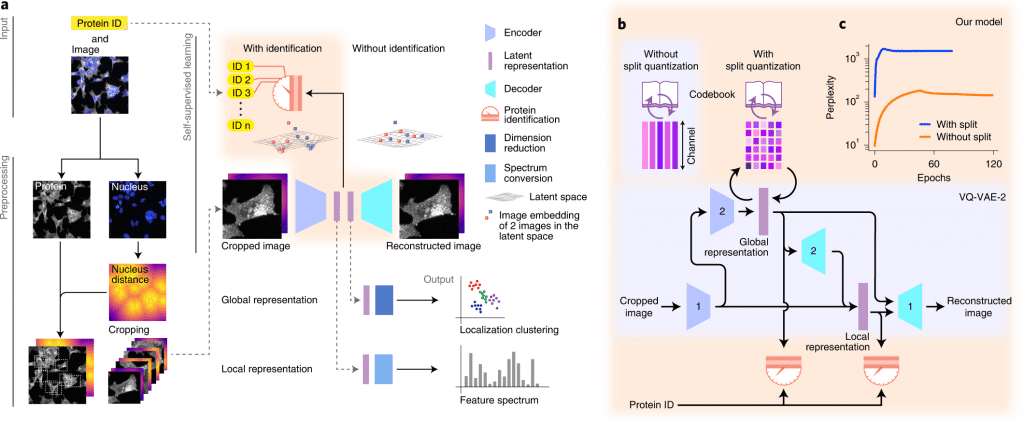
Image Source: https://doi.org/10.1038/s41592-022-01541-z
The results demonstrate that cytoself can cluster proteins into organelles and protein complexes better than previous self-supervised methods. Additionally, it is assessed how each component of this approach contributes to the model’s performance by dissecting the emergent features from which the clustering is derived before interpreting them in the context of the fluorescence images.
As a vital tool for drug screening, drug profiling, and mapping the subcellular localization of proteins, systematic and large-scale microscopy-based cell assays are becoming increasingly important in biological discovery. Human and yeast proteomes can be comprehensively analyzed using large-scale immunofluorescence datasets and endogenous fluorescent tags. The spatial architecture of cells can be systematically mapped by combining such datasets with recent advances in computer vision and deep learning.
There are certain parallels between this situation and the early days of genomics, when the advent of high-throughput, high-fidelity sequencing technologies was accompanied by the development of algorithms for analyzing, comparing, and categorizing these sequences. Despite this, images pose unique challenges to analysis. Microscopy images cannot be compared against a frame of reference (that is, genomes), whereas sequences can. There is no doubt that cells display a wide range of shapes and appearances that reflect a diversity of states.
Modeling and analyzing such rich diversity requires much more skill than, for example, analyzing sequence variability. In addition, most of this diversity is stochastic, making it even more challenging to separate biologically relevant information from irrelevant variance. An image-based screen presents a fundamental computational challenge in obtaining vectorial representations that accurately capture only the biologically relevant information, which can then be quantified, categorized, and interpreted biologically.
In the past, image classification and comparison methods have relied on engineered features like cell size, shape, and texture to quantify different aspects of image content. Despite these features being relevant and interpretable by design, the underlying assumption is that they can be identified and properly quantified to analyze an image.
The recent success of deep learning challenges this assumption. Hand-crafted features cannot compete with learned features that are automatically discovered from the data themselves in a wide range of computer vision tasks, such as image classification. When features are available, the annotation process is typically boot-strapped either by:
- Clustering techniques that do not require supervision, or
- Manual curation and supervised learning.
Supervised approaches involve human annotators inspecting images and marking them with annotations, then training a machine learning model in a supervised manner and applying it to unannotated images.
In addition, models trained on natural images can be reused to learn generic features that can be used to boot-strap supervised training. These approaches are successful, but they are subject to biases since manual annotation imposes its own preconceptions. The ideal algorithm should not rely on human judgment or knowledge but instead synthesize features and analyze images based solely on the images themselves, without making any prior assumptions.
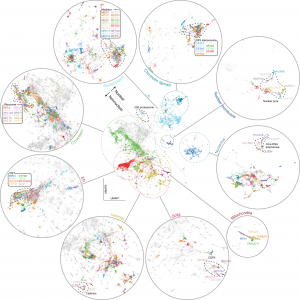
Image Source: https://doi.org/10.1038/s41592-022-01541-z
It is common to train self-supervised models by withholding parts of the data and instructing them to predict them with an auxiliary pretext task. Data sets often contain task-relevant information spread over multiple dimensions. Using a car picture, for instance, we can detect the presence of a vehicle even if many pixels are hidden, perhaps even when half the image has been obscured.
Let’s take a look at a large dataset of real-world object pictures. In order to predict hidden parts from these images, a model must be trained to identify their important characteristics. By capturing the essential features of the images and categorizing them, vectorial representations emerge from pretext tasks.
A deep learning-based approach for self-supervised profiling and clustering of protein subcellular localizations has been developed, validated, and used, called cytoself. The key innovation is a pretext task that helps distinguish microscopy images of that protein from images of other proteins in the dataset by using localization features emerging from different images of the same protein. The cytoself algorithm is demonstrated to be capable of reducing images to feature profiles indicating protein localization, validating its use to forecast protein assignment to organelles and protein complexes, and comparing its performance with previous image enhancement methods.
Data generation has outpaced the ability of humans to annotate it manually. It has already been shown that the abundance of image data has a quality of its own: an increased image dataset has a higher impact on performance than improving the algorithm. In the future, self-supervision will be a powerful tool to handle the large amount of data generated by new instruments, end-to-end automation, and high-throughput imaging-based tests.
Article Sources: Reference Paper | Reference Article
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Srishti Sharma is a consulting Scientific Content Writing Intern at CBIRT. She's currently pursuing M. Tech in Biotechnology from Jaypee Institute of Information Technology. Aspiring researcher, passionate and curious about exploring new scientific methods and scientific writing.





{kind=link}