
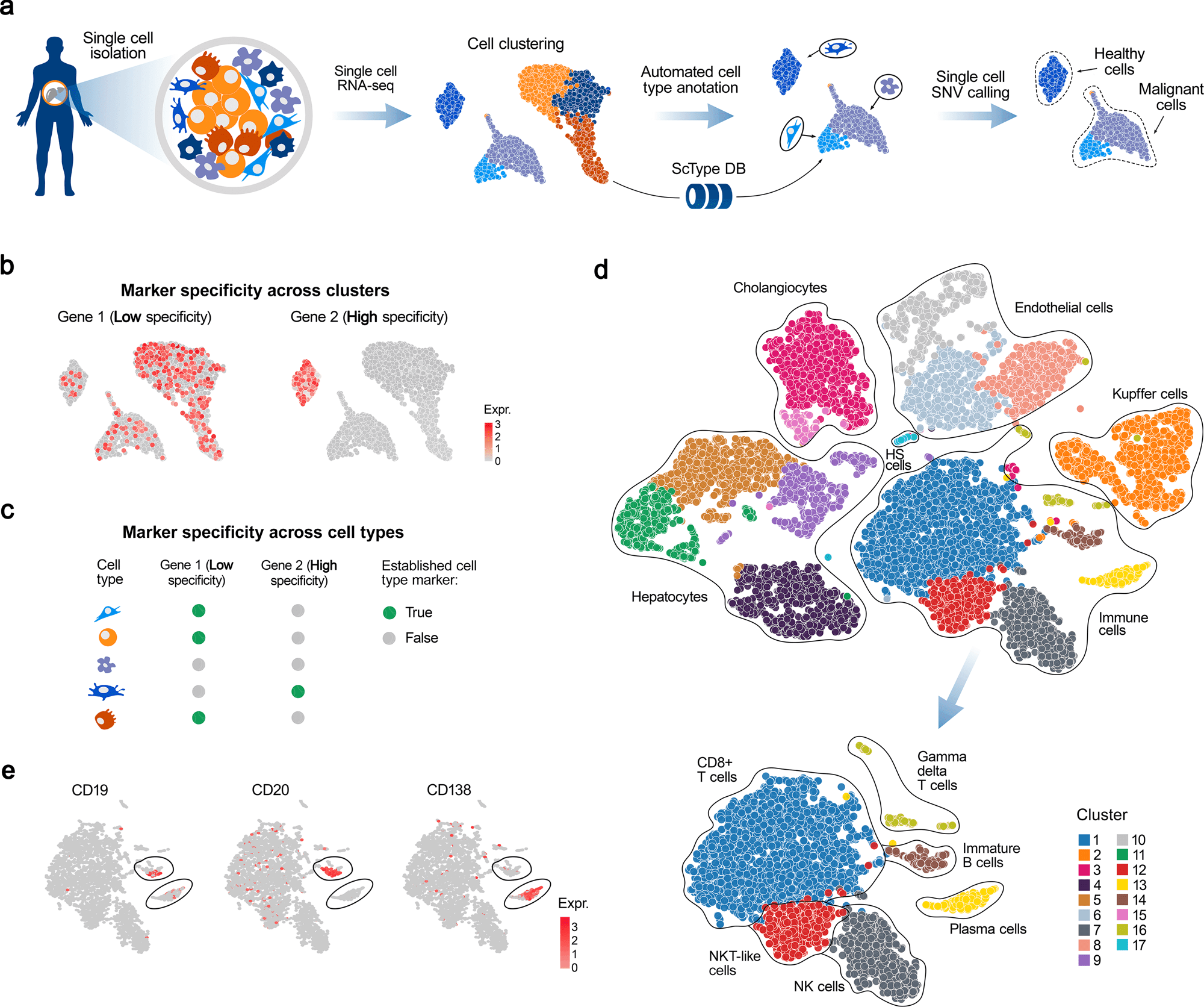
The researchers developed a computational platform ScType, which empowers a completely mechanized and super quick cell-type distinguishing proof-dependent exclusively upon given scRNA-seq information, alongside a thorough cell marker data set as foundation data.
Utilizing six scRNA-seq datasets from different human and mouse tissues, it was shown how ScType gives fair-minded and precise cell type explanations by ensuring the particularity of positive and negative marker qualities across cell bunches and cell types. It was likewise shown how ScType recognizes the sound and threatening cell populations in light of single-cell calling of single-nucleotide variations, making it an adaptable instrument for anti-cancer applications. The generally appropriate strategy is conveyed both as an intuitive web instrument (https://sctype.app) and an open-source R-package.
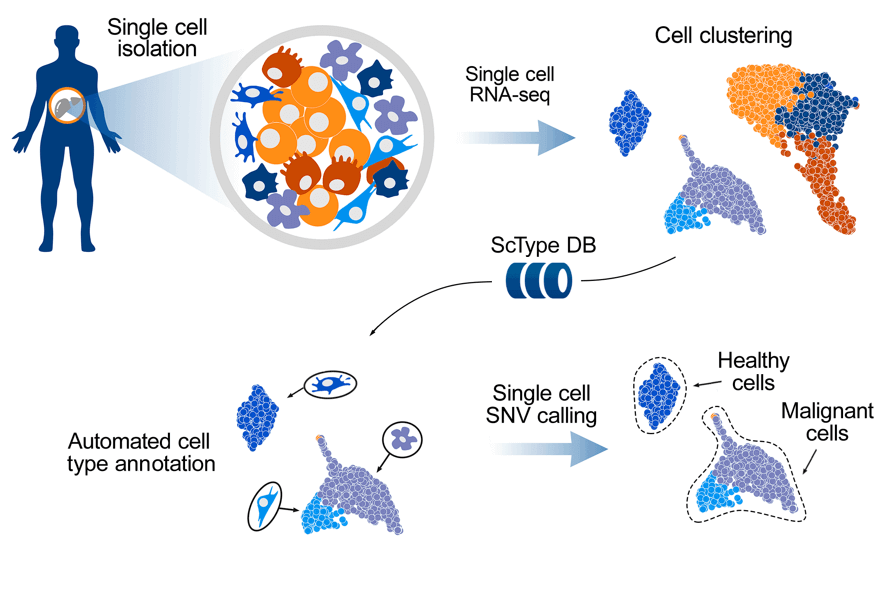
Image Source: Fully-automated Story Source: Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data.
Precise IDs of particular cell types in complex tissue tests are essential for explaining the jobs of cell populations in different organic cycles, counting hematopoiesis, undeveloped and digestive development. Customarily, cell arranging and infinitesimal strategies have been broadly used to detach cell types, followed by atomic profiling of the arranged cells utilizing, for example, mRNA or protein measurements.
Many years of research have prompted a few assortments of cell-explicit highlights, remembering articulation of marker qualities for different tissues that are being utilized to recognize different cell types in new tissue samples. Be that as it may, the whole cycle is physically dreary and testing.
Single-cell RNA sequencing (scRNA-seq) has been laid out as a high-throughput approach to regularly diagram assorted cell populaces in tissue tests and concentrate on different organic cycles in sickness and development. The scRNA-seq innovation has given a unique perspective on different cell types, and it has turned into the driving innovation in enormous scope cell planning undertakings, for example, the Human Cell Atlas.
Distinguishing proof of cell populations frequently depends on an unaided grouping of cells in light of their transcriptomic profiles, followed by group comment utilizing marker qualities that are differentially communicated between the clusters. These marker qualities are then, at that point, physically investigated involving accessible data in writing, or then again, cell marker databases to relegate cell type names to each identified bunch.
Nonetheless, the manual determination of bunch-specific marker combinations is a tedious and mistake-inclined task since the marker qualities are regularly (i) communicated in different cell bunches and (ii) relate to different cell types. Likewise, the articulation of negative marker qualities, which give proof against a cell being of a specific kind, ought to be fused into the cell-type distinguishing proof cycle.
The cell comment strategy is further confounded by the absence of organized cell marker information bases that incorporate both known and all over again certain and negative markers to explain cell types with certainty. For instance, the choice of CD44 as marker quality might think twice about the precision of cell explanation as CD44 is communicated in different safe cell populations. One more famous methodology for cell-type tasks is to use a reference dataset, an assortment of already annotated cell types in single-cell information, to prepare an arrangement algorithm and to apply it to new single-cell datasets. In any case, such administered approaches expect the reference and new datasets to look like one another, which frequently represents an issue in scRNA-seq studies. One significant use of single-cell portrayal is to configure customized medicines that specifically target harmful cell types in a patient-determined example while staying away from serious hindrance and poisonous consequences for sound cells. In tumors and other complex sicknesses, monotherapy obstruction frequently arises.
What’s more, it requires multi-drug co-hindrance of different infections and opposition driving cell populations. It was as of late illustrated how our far-reaching ScType marker data set helped an AI-directed recognizable proof of customized drug mix treatments for patients with unmanageable intense myeloid leukemia (AML), which prompted synergistic co-hindrance of leukemic cell subpopulations that arose in different phases of the infection pathogenesis and treatment regimens. These disease-specific and patient-explicit mixes were demonstrated to be generally less poisonous to lymphoid cells (non-dangerous cells in the AML case), in this manner expanding their probability for clinical interpretation. In any case, how to accurately recognize numerous harmful and non-dangerous cell populations for designated medicines remains a translational challenge and requires both methodical and profoundly special strategies that are relevant to different sicknesses and tissue types. In numerous biomedical applications, reference single-cell information and cell type comments are not accessible. Moreover, the cell population distinguishing proof should be done separately for every quiet test.
To solve these difficulties, a computational ScType stage (marker information base and cell-type recognizable proof calculation) was fostered, which requires just a solitary scRNA-seq dataset for an exact and unaided cell-type explanation. The fair-minded at this point specific cell-type comment is accomplished by accumulating the biggest data set of laid out cell-explicit markers (ScType data set) and by guaranteeing the particularity of marker qualities across both the cell groups and cell-types (ScType explicitness score).
A precise benchmarking of ScType and related strategies was completed across six scRNA-seq datasets from four human and two mouse tissues and showed that the ScType stage accurately annotated a sum of 72 out of 73 cell-types (98.6% precision), including 8 recently re-annotated cell types that were inaccurately or non-explicitly explained in the first investigations. Moreover, ScType executes a solitary cell single-nucleotide variation (SNV) calling choice to recognize threatening and non-dangerous cells, exemplified here utilizing scRNA-seq information from the AML patient test. This contextual investigation exhibits how the ScType stage can be used for anti-cancer applications. For example, information-driven identification of leukemic cell populations toward customized and specific treatment choice. ScType step is carried out as an open-source and intelligent web device (https://sctype.app), associated with the ScType marker data set, to empower super quick and completely mechanized cell-type explanation in a broad scope of biomedical applications.
The ScType further develops explanations of cell types dependent exclusively upon a piece of given scRNA-seq information. The presentation of ScType was initially explored by re-examining a distributed scRNA-seq investigation of human liver cells. Utilizing just the crude scRNA-seq information from the liver chartbook dataset, ScType consequently distinguished 17 groups and accurately appointed them to 11 distinguished liver-related cell types that were physically clarified in the first review. This exhibits the advantages of the complete marker information bases. Furthermore, the exactness of the completely computerized comment process. Also, ScType had the option to consequently recognize between two intently related cell populaces of B-cells (youthful also, plasma B cells) that were not separated in the first manuscript. This isolation among juvenile and plasma B cells was done because of the positive and negative data in the ScType data set between the plasma cells that don’t communicate with normal B-cell markers, like CD19 and CD20, yet instead, they express CD138.
Orderly benchmarking of ScType in human and mouse scRNA-seq informational collections
To explore the more extensive appropriateness of the computerized strategy, it was next benchmarked the exhibition of ScType as far as its capacity to consequently dole out cell-types compared to the cell-type explanations given by the first creators of four additional distributed scRNA-seq studies. It was further studied that all the six datasets looked at ScType execution against the other ongoing cell-type comment strategies as far as their precision and running time. The RNA-seq datasets utilized in the benchmarking began from different tissues, including the human liver, pancreas, peripheral blood mononuclear cells (PBMCs), brain, and mouse lung and retina samples. These scRNA-seq datasets encouraged the researchers to study the performance of ScType and the connected techniques with regards to different sequencing stages, tissue types, and life forms.
ScType uses both positive and negative markers for the cell type explanation. As an extraordinary element, ScType acknowledges and makes the utilization of positive markers, yet in addition, negative marker qualities, i.e., markers that are not relied upon to be communicated in a specific cell type, with the plan to separate between firmly related cell types. It is observed that a similar quality can be a positive marker for one cell type and a negative marker for another cell type.
Single-cell SNV calling recognizes the sound and harmful cell types. To empower hereditary examinations in disease applications, there is further a possibility for single nucleotide variety (SNV) called straightforwardly from the scRNA-seq information. For instance, it was re-investigated that the scRNA-seq transcriptome profile and cell-type organization of an AML patient test from the new study utilized the ScType stage.
Story Source: Ianevski, A., Giri, A.K. & Aittokallio, T. Fully-automated and ultra-fast cell-type identification using specific marker combinations from single-cell transcriptomic data. Nat Commun 13, 1246 (2022). https://doi.org/10.1038/s41467-022-28803-w
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.





{kind=link}