
Scientists from the Genome Institute of Singapore and A*STAR Singapore have developed a neural network-based method that implements a multi-instance learning framework, “m6Anet,” to accurately detect the RNA modification m6A from genomic data.
Around 150 types of RNA modifications are currently known, of which m6A methylation is noteworthy for its role in diseases. These modifications add to the complexity of analyzing transcriptome data. These modifications are difficult to detect with the current approaches available to researchers, which lack single-molecule resolution. However, Nanopore direct RNA sequencing can capture RNA modification data for the RNA molecule. This enables the detection of RNA modifications using supervised machine learning-based techniques using nanopore sequencing data. The authors train a neural network, m6Anet, with ample data from Nanopore direct RNA sequencing and implement a multi-instance learning approach to detect m6A. The method is shown to outperform current computational methods, and its accuracy is comparable with laboratory experiments. Without retraining model parameters, m6Anet generalizes to different cell lines and other species accurately. m6Anet is able to capture read-level stoichiometry, thereby enabling approximation of differences in modification rates.
RNA modifications and why we need to detect m6A
RNA modifications can lead to alterations in the genetic information of the organism. They were first discovered in the 1950s. Currently, more than 150 modifications have been identified, with m6A, the main internal methylation on mammalian mRNA, being one of the most commonly found RNA modifications. This modification, found mostly at the consensus motif DRACH, impacts RNA structure, splicing, stability as well as translation. In humans, this modification has been shown to play significant roles in cancer and other diseases.
Experimental approaches used for detecting RNA modifications are broadly classified into immunoprecipitation-based methods, chemical-based detection methods, and specific enzyme-based methods. These approaches have resulted in numerous methods that yield a transcriptome-wide map of RNA modification sites. However, due to the lack of available antibodies for specific modifications, the scope of these methods is limited.
The Nanopore direct RNA sequencing technique enables the detection of modification sites without limitations and hence is a perfect candidate to incorporate into the framework for the computational detection of RNA modifications.
Multi-instance learning: the game-changer
Comparative approaches for detecting RNA modifications typically detect m6A sites in comparison with samples that lack m6A modifications. These methods do not require training data. Comparative methods like Tombo, DRUMMER, nanoDOC, Nanocompore, ELIGOS, xPore, and Yanocomp all use comparative approaches to accurately detect m6A modification sites. However, the requirement of m6A-free data control samples is a limiting factor as it requires silencing writer genes.
Supervised machine learning-based approaches are not threatened by these limitations. Supervised detection of modification sites is a classification problem that requires training the classifier with labels obtained from data of synthetically modified RNA samples or data from other experimental methods. While supervised methods do not need modification-free sample data for comparison, they are less accurate than comparative methods. Supervised learning-based methods are posed with the challenge of missing training labels for each individual reads. This is a multi-instance learning problem, and m6Anet has been developed by training a multi-instance learning classifier with single-read level data, a true game-changer.
Multi-instance learning (MIL) is a fairly new learning paradigm as compared to traditional supervised learning. This is typically applied where the data is somewhat ambiguously labeled, and instead of training the algorithm with input/label pairs, labels are assigned to bags or sets of inputs. The labels are binary, and the assumption in the MIL paradigm is that every positive bag has at least one positive input.
In the context of m6A modification detection, m6Anet takes direct RNA sequencing data and transforms it into a MIL problem. The model incorporates modified and unmodified RNA data and outputs the m6A modification probability at any particular site for all DRACH five-mers represented in the training data. The main difference between m6Anet and other supervised methods lies in the fact that the algorithm is trained to learn high-dimensional representations of individual reads from every candidate position and then aggregates the information to generate probabilities for m6A sites more accurately.
The all-encompassing technique in m6A detection
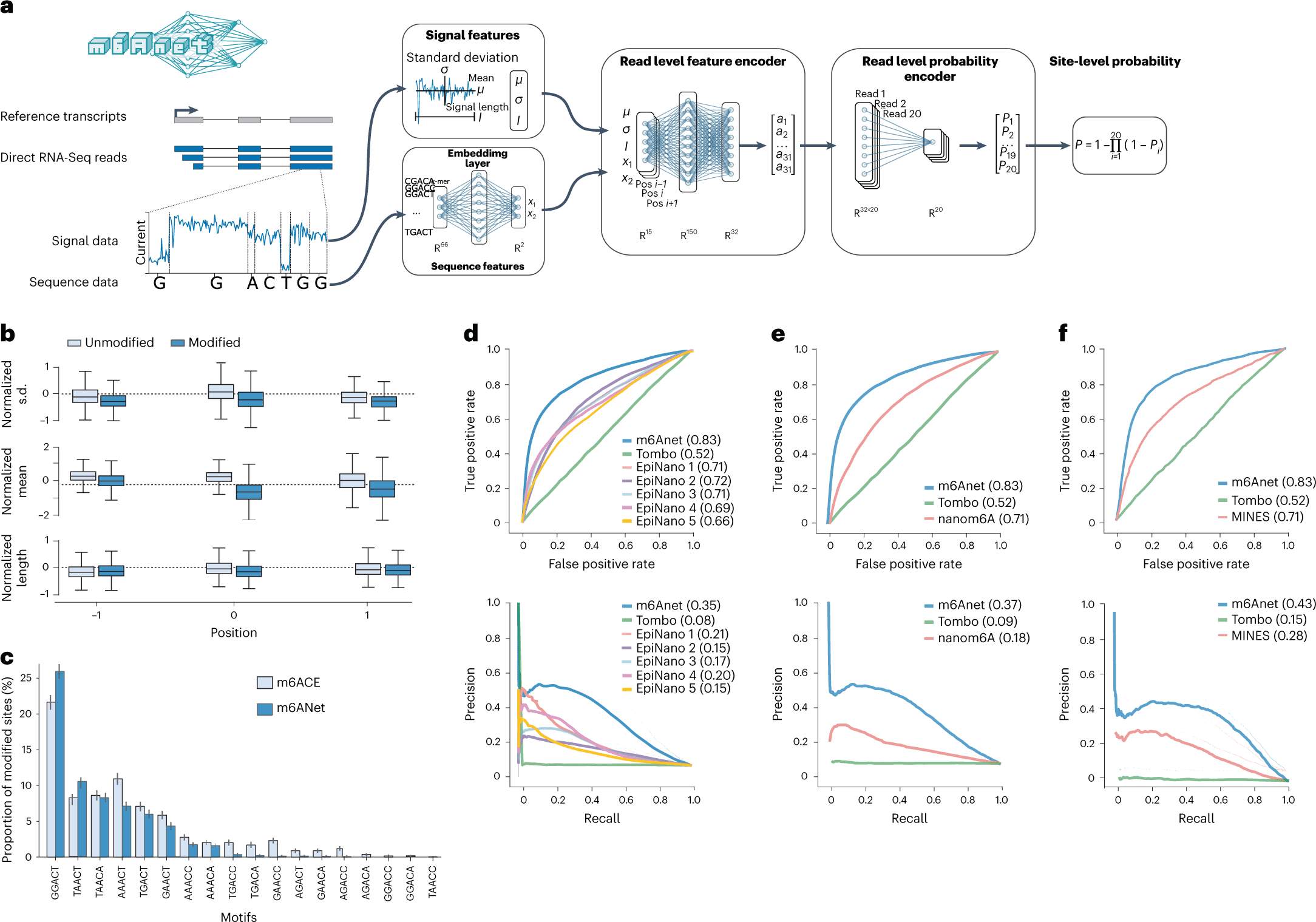
The method, m6Anet, is composed of two modules, a read-level encoder and a pooling layer, which are jointly optimized. The encoder generates probability for each read modification by incorporating signal and sequence features from each read that is transformed into a high-dimensional representation. The pooling layer pools the read-level probability to generate probability estimates for a site being modified.
The following figure illustrates the model modules.
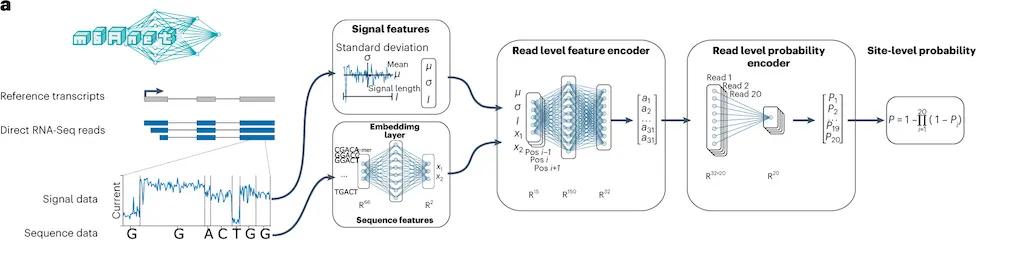
Image source: https://doi.org/10.1038/s41592-022-01666-1
- m6Anet identifies methylated positions m6A accurately.
- m6Anet has been shown to detect m6A sites more efficiently and accurately compared to other methods.
- m6Anet can be applied to different cell lines other than the cell line used and other species. The method was applied to two different human cell lines, HEK293T and Hct116 datasets and the Arabidopsis dataset.
- The method is shown to achieve high precision with top-predicted sites.
- m6Anet generates single-molecule m6A predictions.
- m6Anet captures the m6A stoichiometry, which can be used for comparing sites within one sample.
Conclusion
The authors developed the m6Anet software for detecting m6A modifications from nanopore direct RNA sequencing data. The method implements a neural-network-based MIL algorithm that uses signal and sequence features from the direct sequencing data to generate probability estimates for the modification of sites using a read-level encoder and a pooling layer. Apart from detecting m6A sites, the method can also capture m6A stoichiometry, which is a remarkable feat as this will open up new frontiers in molecular biology research. The method is highly accurate and efficient and can be generalized to different cell lines and species without retraining model parameters. The accurate detection of m6A sites accurately will certainly aid in drug design and delivery for diseases wherein m6A RNA modification has a significant role.
Article Source: Reference Paper | Reference Article
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





{kind=link}