
Scientists from the University of Hamburg, Germany, have developed a novel methodology, DiNiro, for unraveling differential regulatory disease mechanisms from single-cell RNA-seq data. Computational tools for analyzing differential gene expression profiles and differential pathway expression have been previously developed. DiNiro is the first known method based on single-cell data for analyzing differential regulatory disease mechanisms. DiNiro has been developed not just to predict but also to explain differential cellular gene expression programs. DiNiro predicts and reports these differential mechanisms as small,easy-to-understand transcriptional regulatory network modules using copulas. The authors also apply the method to disease case studies, including SARS-CoV-2 and autism.
Differential co-expression analysis and the need for DiNiro
The evolution of single-cell RNA sequencing (scRNAseq) technology has resulted in a plethora of methods for studying and analyzing cellular heterogeneity and gene expression variability at the single-cell level resolution. However, the amount of data being generated by scRNA–seq technologies is far more than the methods being produced for their analyses. The need for more advanced computational methods for analyzing such data is far more than improving the current standard methods to gain better insights into the underlying biological mechanisms. Differential single-cell enrichment analysis of scRNAseq data could be a storehouse of unexplored and significant information that could reveal crucial insights.
Single-cell RNA sequencing data analysis typically involves two components of pre-processing data and downstream analysis. Numerous methods have been developed for both of these steps. Several tools and packages exist to facilitate this downstream analysis. Differential gene network enrichment analysis using scRNAseq data could yield novel regulatory phenotypes or disease mechanisms that could not be otherwise determined with co-expression and differential expression analysis.
With differential co-expression analysis, it is possible to identify gene pairs comprising transcription factors (TF) and their target genes (TG) which show distinct co-expression patterns across cell clusters. Co-expressing genes with an appreciable co-expression association could result in a network. Gene regulatory networks (GRN) inference is mostly used in the co-expression analysis for genes. GRNs are typically modeled as directed graphs with TF and TG as nodes and activation and repression as directed edges between them. Single-cell-based GRN (scGRN) analysis is essential for understanding cellular mechanisms driving various processes. However, constructing scGRN from scRNAseq data has met with limited success in terms of accuracy, as seen by the benchmarking studies.
Previous methods such as PPCOR, PIDC, and SCENIC identify gene co-expression modules and directed networks. However, no tool exists for differential scGRN reconstruction. Thus, the authors developed DiNiro.
DiNiro framework overview
The framework is developed to compare two single-cell samples/groups/conditions using gene regulatory networks and copulas at the gene level. DiNiro conducts a complete differential co-expression analysis procedure and outputs the differentially regulated gene modules across the samples. The following figure illustrates the basic methodology involved in the framework.
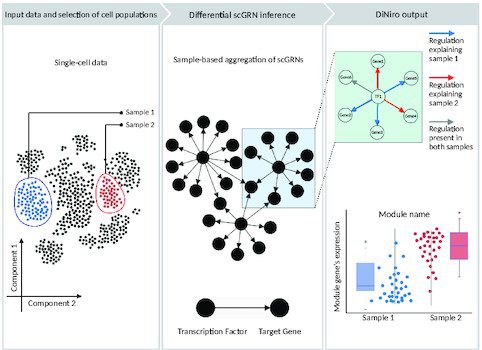
Image source: https://doi.org/10.1093/nargab/lqad018
Workflow
- The analysis begins with selecting two samples from input single-cell data.
- GRNBoost2 is used for inferring the scGRNsfrom the sub-samples.
- This identifies potential TGs for every TF from the co-expression data.
- Sub-sample-based scGRNs are denoised and aligned for constructing one sample-based scGRN.
- Two scGRNs are fused to form TFxTG adjacency matrix, which acts as the input for the copula-based method for identifying differential co-expression modules.
The following figure illustrates the DiNiro workflow:
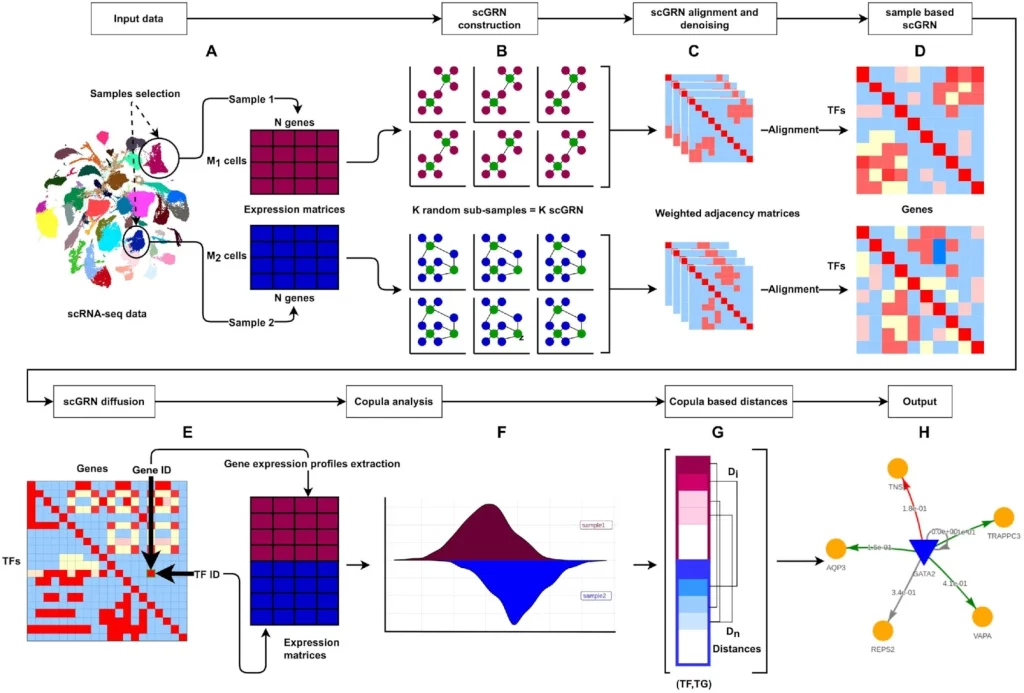
Image source: https://doi.org/10.1093/nargab/lqad018
The authors performed a three-level validation analysis to evaluate the performance and robustness of DiNiro.
- Benchmarked DiNiro against state-of-the-art GRN inference tools.
- Performance evaluation was done using simulated data with already known gene regulatory networks revealing DiNiro’s power in terms of lowering false positives.
- DiNiro was applied to three real-word scRNseq or snRNA-seq datasets, which led to novel discoveries.
Real-world case studies
The authors illustrate the capabilities of DiNiro by applying it to single-cell datasets from three real-world scenarios. First, they examine molecular mechanisms of T-cell fate after chronic viral infection. Second, they examine the interactome of host cells infected with SARS-CoV-2 for cellular dynamics. Third, they examine pathological networks in brain cell subtypes that are susceptible to changes in autism. These resulted in the discovery of novel networks underlying these diseases.
Conclusion
The authors have developed a novel methodology and framework for the analysis of the differential regulatory disease mechanism, DiNiro. The method has been benchmarked against state-of-the-art GRN inference-based methods and has been shown to yield better performance and accuracy in predicting co-expression gene modules across samples. The methodology involves denoising the input data at the pre-processing step to deal with stochastic transcriptional noise and other biological noise sources. The main method part for identifying the gene pair of TFs and their TGs and for generating the modules involves the use of copulas. The authors have also revealed the power of DiNiro by applying it to three real-world scenarios and discovered novel networks involved in underlying disease mechanisms. The authors anticipate the future use of this methodology to single-cell OMICS analysis for comparing between species. DiNiro is clearly a groundbreaking method for facilitating not only the identification of novel networks and gene modules but also for drug design, diagnostics, and therapeutic developments. With the aid of this framework, single-cell RNA sequencing data-based gene co-expression analysis will open doors to many novel discoveries that will further our understanding of fundamental biological mechanisms at the single-cell level.
Article Source: Reference Paper | DiNiro: Platform
Learn More:




Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





{kind=link}