
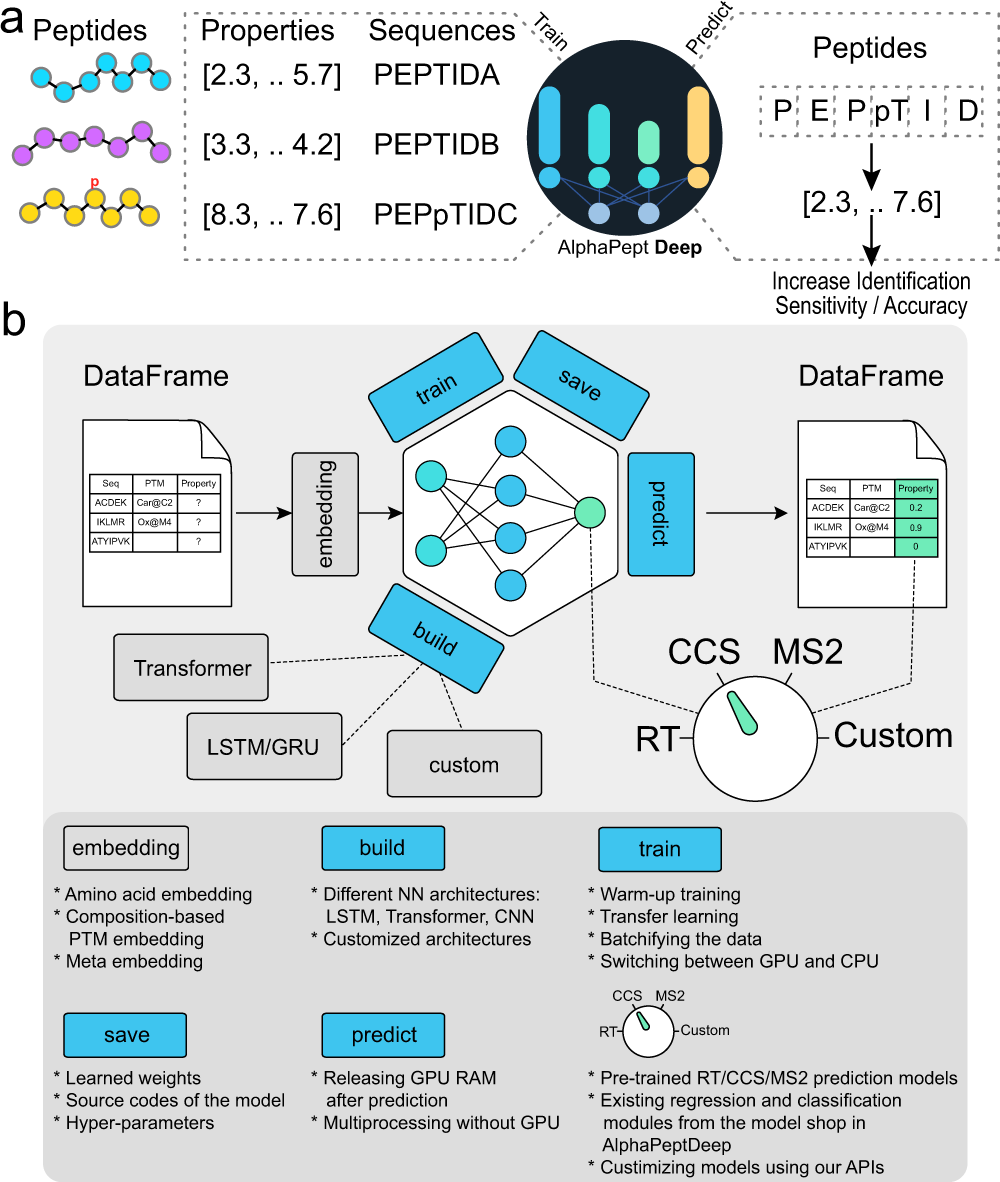
A research team led by Professor Matthias Mann from the Max Planck Institute of Biochemistry has developed AlphaPeptDeep. AlphaPeptDeep is a Python framework built on the PyTorch deep learning library that can learn and predict peptide properties. AlphaPeptDeep additionally includes a plug-in capability that allows non-experts to develop models with just a few lines of code. AlphaPeptDeep may also indicate additional sequence-based features, as demonstrated with a Human Leukocyte Antigen (HLA) peptide prediction model.
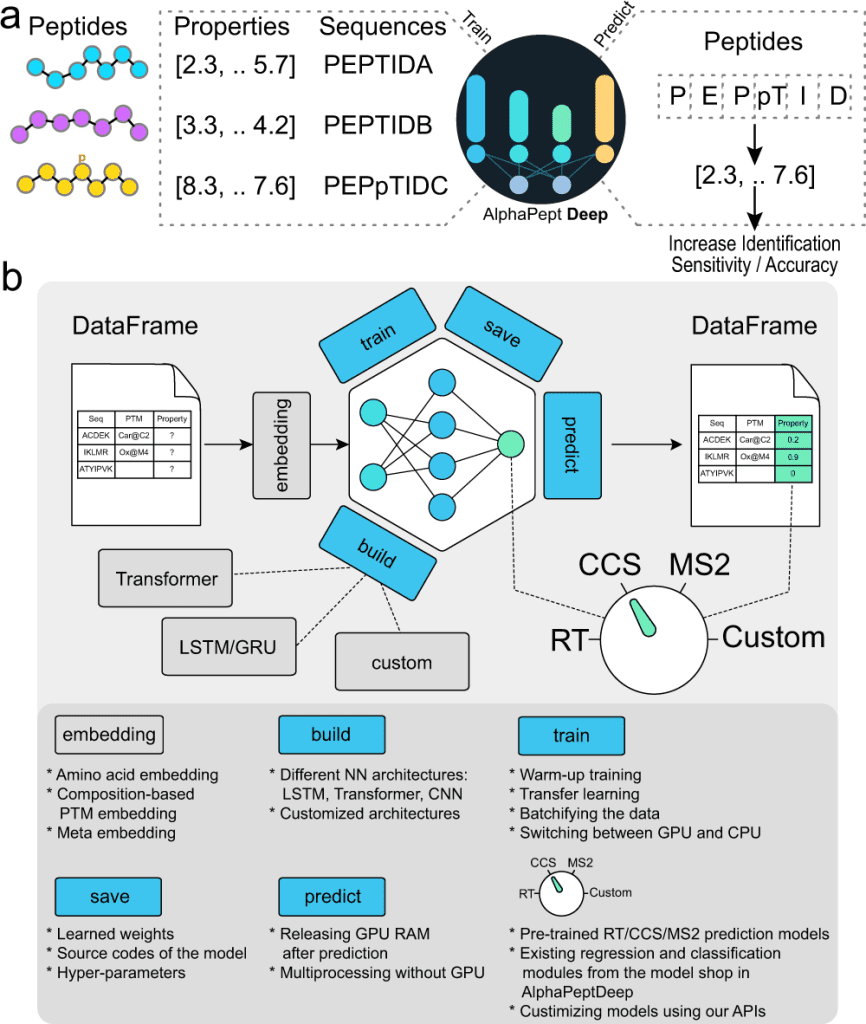
Image Source: https://doi.org/10.1038/s41467-022-34904-3
Mass spectrometry is a technique to determine the mass of ionized molecules and biomolecules and is increasingly used for protein identification and quantification. Exploration of diverse elements of the proteome is possible by selecting the proper mix for sample preparation, experimental apparatus, and data processing. The most exhaustive approach for quantitative profiling of proteins, their interactions, and changes is mass spectrometry (MS)-based proteomics.
Mass spectrometry-based proteomics seeks to gain an unbiased assessment of all the proteins present in a particular system. This difficult analytical process necessitates sophisticated liquid chromatography/mass spectrometry (LC/MS) instruments downstream with bioinformatic analysis pipelines. Deep learning (DL) techniques have grown in potency over the last decade and are becoming increasingly valuable for MS-based proteomics. In proteomics, the identification includes matching fragmentation spectra (MS2) and other features to a set of peptides. Bioinformatics can now predict peptide properties for every amino acid sequence and compare them to real observed data. This can significantly boost statistical confidence in peptide identifications.
The authors of this study set out to develop AlphaPeptDeep, a comprehensive and simple-to-use framework. AlphaPeptDeep makes use of PyTorch, one of the most prominent DL libraries. AlphaPeptDeep includes pre-trained models for predicting MS2 intensity, RT values, and collisional cross sections (CCS) of peptide sequences or proteomes. It also handles peptides with post-translational modifications (PTMs), including unknown ones with user-specified chemical compositions. AlphaPeptDeep makes considerable use of transfer learning, which significantly reduces the amount of training data necessary.
AlphaPeptDeep, in conjunction with its built-in Percolator implementation, significantly improves peptide identification performance for data-dependent acquisition on complex samples such as HLA peptides. The study report also shows how AlphaPeptDeep can be easily used to build and train models for various peptide properties.
Major Findings
AlphaPeptDeep, a deep learning framework, integrates various functionalities for training, transferring, and employing models for peptide property prediction. AlphaBase is an infrastructure package that was used for building AlphaPeptDeep, which includes numerous features required for proteins, peptides, PTMs, and spectrum libraries. Based on these features, Zeng et al. created MS2, RT, and CCS models that allowed the prediction of a wide range of post-translational modifications. These models improved the detection of HLA peptides, for example, not just in standard search but also in open search. A unique module called the ‘model shop’ offers generic models that customers may utilize to create new ones with just a few lines of code. An HLA prediction model based on the model shop was also performed to predict whether a peptide sequence is an HLA peptide.
The HLA spectral libraries were directly created from the complete human proteome using the HLA model, MS2, RT, and CCS models in AlphaPeptDeep. The libraries used in the current study outperformed existing DDA and DIA operations.
Limitations
Although AlphaPeptDeep is solid and simple, standard machine learning concerns such as overfitting in the framework prevail. Users must partition the data, train and test the models on various sets, and experiment with different hyperparameters is still required. The differences in batch sizes and learning rates may also affect model training. On the other hand, the model store provides baseline models for any property prediction task. AlphaPeptDeep is expected to make it easier for non-AI specialists to develop their models, simulated from scratch or in tandem with other pre-trained models.
Conclusions
Peptide property prediction can be employed to improve every step of the computational proteomics workflow. Aside from specific qualities of interest in MS-based proteomics, DL models may be used to tackle a problem where the peptide property is an inherent function of an amino acid sequence that is demonstrated by successfully predicting probable HLA peptides. As a result, the AlphaPeptDeep framework will be a valuable deep-learning resource for proteomics if adequate and accurate training data are available.
Article Source: Reference Paper | Code: AlphaPeptDeep | AlphaBase
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Shwetha is a consulting scientific content writing intern at CBIRT. She has completed her Master’s in biotechnology at the Indian Institute of Technology, Hyderabad, with nearly two years of research experience in cellular biology and cell signaling. She is passionate about science communication, cancer biology, and everything that strikes her curiosity!





{kind=link}