
All organisms depend on protein-protein interactions (PPIs) for cellular and biological activities. This paper introduces a novel method for building peptide binders for “undruggable” protein-protein interactions. A machine learning approach generates different candidate peptides from natural binding regions from protein partners. Molecular docking and in-vitro microchip-based binding assays filter the peptides. The research confirms and evaluates the technique on calcineurin and finds several peptides that interfere with its substrate binding. Protein-protein interaction modulators are found using protein interaction and sequence databases, generative modeling, molecular docking, and interaction tests.
Targeting Protein-Protein Interactions and Calcineurin
Drug development targets protein-protein interfaces because they are implicated in many disease pathways where therapeutic intervention will help many people. Short amino acid sequences called peptide binders are intended to attach to certain target proteins, such as “undruggable” protein-protein interfaces. Traditional small-molecule medications often fail to penetrate these protein-protein interfaces, but they may be targeted by peptide binders, which can alter their action. Therefore, the design of peptide binders is a viable technique for the development of novel therapeutics for illnesses involving these kinds of proteins, such as cancer and neurological disorders. They can mimic the binding site of one of the partners and are estimated to cover up to 40% of protein-protein interactions (PPIs).
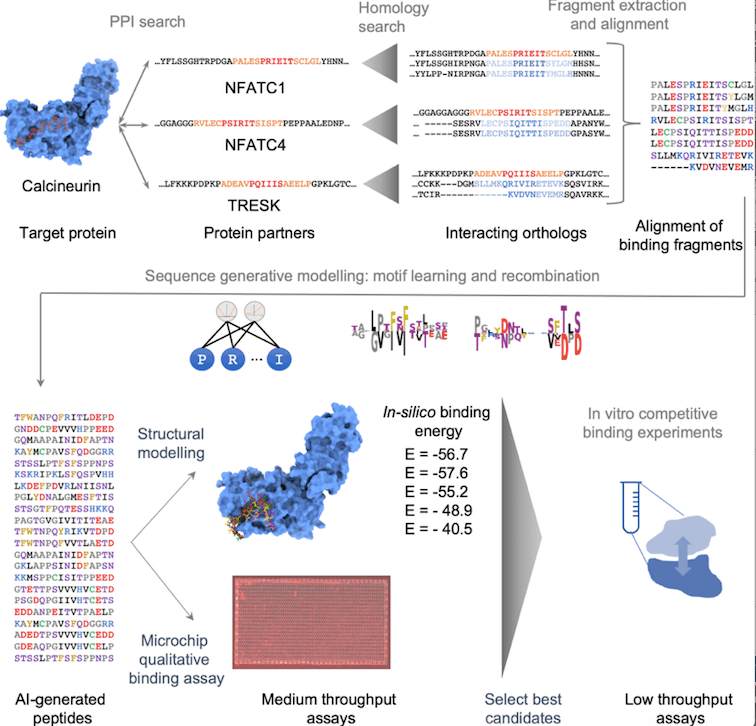
The enormous amount of potential sequences makes peptide discovery difficult. Using protein-protein complex architectures to determine initial peptide sequences helps. Machine learning sequence generative models (SGM) can now understand protein biophysical limitations and explore sequence space. Training these models needs a vast and diversified collection of evolutionary-related sequences. The target protein-protein interaction (PPI) may be conserved in a few animals or mediated by highly conserved short linear motifs, making peptide design difficult. An integrative peptide design protocol with four steps is proposed to overcome these limitations: construction of multiple alignments of putatively binding fragments, training and validation of an SGM, generation of a library of candidate peptide sequences, and filtering of the library through in-silico docking and in-vitro binding assay.
Calcineurin (Cn) is a protein phosphatase involved in essential physiological functions. Cn inhibitors that are currently on the market have undesirable side effects, thus, peptide-based modulators that prevent Cn from attaching to its substrates while keeping its catalytic site may be preferable. Cn’s signaling network has been thoroughly investigated, yielding accurate data on its many protein substrates and binding locations, making it an appropriate case study.
Calcineurin Signaling Network Study
Calcineurin (Cn) is a calcium-dependent phosphatase that dephosphorylates serine and threonine residues to modulate the activity of its substrates, including the NFAT transcription factors. Cn is a heterodimer with catalytic and regulatory domains that are activated by calcium chelation and interaction with calmodulin. At least 29 and 38 substrates have been found in humans and yeast, respectively, for the Cn signaling network. The ScanNet web server was used to predict the binding sites of intrinsically disordered proteins in order to identify substrate-binding sites. There are two substrate binding sites that recognize two SLiMs (PxIxIT and LxVP). The Cn-NFAT interaction is intended to take place only at high calcium concentrations, and earlier research has created peptides (PVIVIT and its variants) that may inhibit Cn-substrate binding and dephosphorylation. This study attempted to develop alternative peptides with increased affinity and specificity since most earlier peptide discoveries were conducted by the experimental screening of sequences near NFAT-derived peptides without investigating the huge array of alternate substrate motif alternatives.
Design Protocol for Peptides
The proposed design protocol consists of four major steps:
- Curation of known Calcineurin-binding fragments
- Data augmentation via homology search
- Peptide library design utilizing a sequence generative model
- In-silico and in-vitro screening followed by quantitative binding assay
The first step begins with a list of 67 known protein substrates and the PxIxIT segments that belong to them. The second phase is data enrichment by homology search and statistical filtering. In the third stage, a compositional Restricted Boltzmann Machine is trained and utilized to build a diversified library of potential peptide sequences. In the last stage, the binding strength of the peptides is calculated in-silico and assessed by in-vitro experiments, and the most promising peptides are chosen for further characterization.
The homology search yielded a multiple alignment of 1886 fragments with significant amino acid preferences at certain places. Visualization revealed pieces primarily clustered by gene and taxon, with central SLIMs being more varied. Conservation patterns beyond the six classic SLIM sites indicated different binding conformations and unique binding energy repartition. Recombining these diverse sequences may increase binding relative to natural equivalents and allow substrate-specific competition.
Generative modeling is an unsupervised learning technique that trains a parametric probability distribution PΘ(S) over the sequence space. It assigns high PΘ(S) values to observable sequences and low values elsewhere, representing the fitness function of evolution. The inductive bias over the sequence space is determined by choice of functional form PΘ(S). Compositional Restricted Boltzmann Machines (cRBM) were employed as the functional form P in this study (S). The cRBM is a robust inductive bias for protein sequence modeling since it incorporates high-order epistatic interaction components and is simpler to comprehend than other models. Multiple fragment alignments were used to train the cRBM, and the best model was chosen based on performance and interpretability. The model’s validity was confirmed by comparing its log-probability function to extensive mutational scans of binding affinity, which revealed a high correlation between certain peptides.
Using the FP (Fluorescence Polarisation) competition assay, it was found that seven out of ten synthetic peptides and three out of four natural peptides were able to compete with PVIVIT for Cn binding, with IC50 values ranging from 1.17M to 250M. The fragment of the human AKAP79 gene with an IC50 of 17.5 M was the second-best peptide. The best synthetic peptide was produced by the rational recombination of several peptides and had an IC50 value of 14 M. All peptides had a motif similar to PxIxIT, however, this was not required for binding. The CAPN11-related peptides failed to compete with PVIVIT, potentially due to different binding or dependency on post-translational modifications.
Limitations
In terms of SGM training, the protocol has limits since it needs a wide collection of sequences. It may not be applicable if the interaction is not well conserved or if several natural binders have not been identified. There is no assurance that all sequences in the multiple sequence alignment used for training bind the target. Due to the fact that peptides are chemically bound to the chip instead of circulating freely in solution, the microchip qualitative binding test may produce a high rate of false negatives.
Conclusion
The suggested technique in this study provides a unique approach to creating peptides that target particular protein binding sites. It involves identifying native partners and their interacting fragments using experimental data and homology search, training a sequence generative model, filtering the in-silico peptides using a cost-effective and medium-throughput approach, and determining the ability of the most promising peptides to inhibit protein-protein interactions. The technique was applied to the Cn-PxIxIT complex, and seven out of ten engineered peptides and three out of four natural peptides bound the target binding site effectively. A natural peptide with a C-terminal proline-rich motif and a recombinant peptide with six mutations were the most effective peptides. These peptides serve as a foundation for the future development of peptide design via in-vitro mutagenesis or transfer learning.
Article Source: Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Sejal is a consulting scientific writing intern at CBIRT. She is an undergraduate student of the Department of Biotechnology at the Indian Institute of Technology, Kharagpur. She is an avid reader, and her logical and analytical skills are an asset to any research organization.





{kind=link}