
To filter promising drug candidates early in the drug discovery process, it is imperative to accurately predict how well a protein and a potential drug molecule (ligand) will bind together. Deep learning methods are increasingly being incorporated into drug discovery, but they struggle to be as versatile as conventional methods due to their suboptimal abilities in extrapolating previously learned information onto new scenarios. Hence, researchers from the University of Edinburgh have attempted to understand what these models are learning from protein and ligand information. They used a deep learning model that relies on sequential and structural information on kinase datasets. Their findings indicate that the deep learning model is much more influenced by ligand information than by protein information.
The Use of AI in Drug Discovery
Predicting the binding affinity between a protein and a ligand helps in the quicker identification of appropriate drug candidates without the aid of time-consuming experiments. Many methods have been developed for predicting the binding strength between proteins and ligands, but all of them are accompanied by limitations, especially when it comes to a large number of compounds. Methods utilizing computer simulations are often inaccurate, and other accurate methods take up a huge amount of computational power. Recently, scientists have included machine learning methods in drug discovery to enhance binding strength prediction.
But even machine learning models struggle with a drawback. They fail to generalize the information they learned during their training and perform suboptimally when presented with new scenarios. To understand how exactly these models work and what improvements could be incorporated into them, researchers used a deep learning model on kinase datasets that takes protein and ligand information as input and analyze the factors on which the results depend.
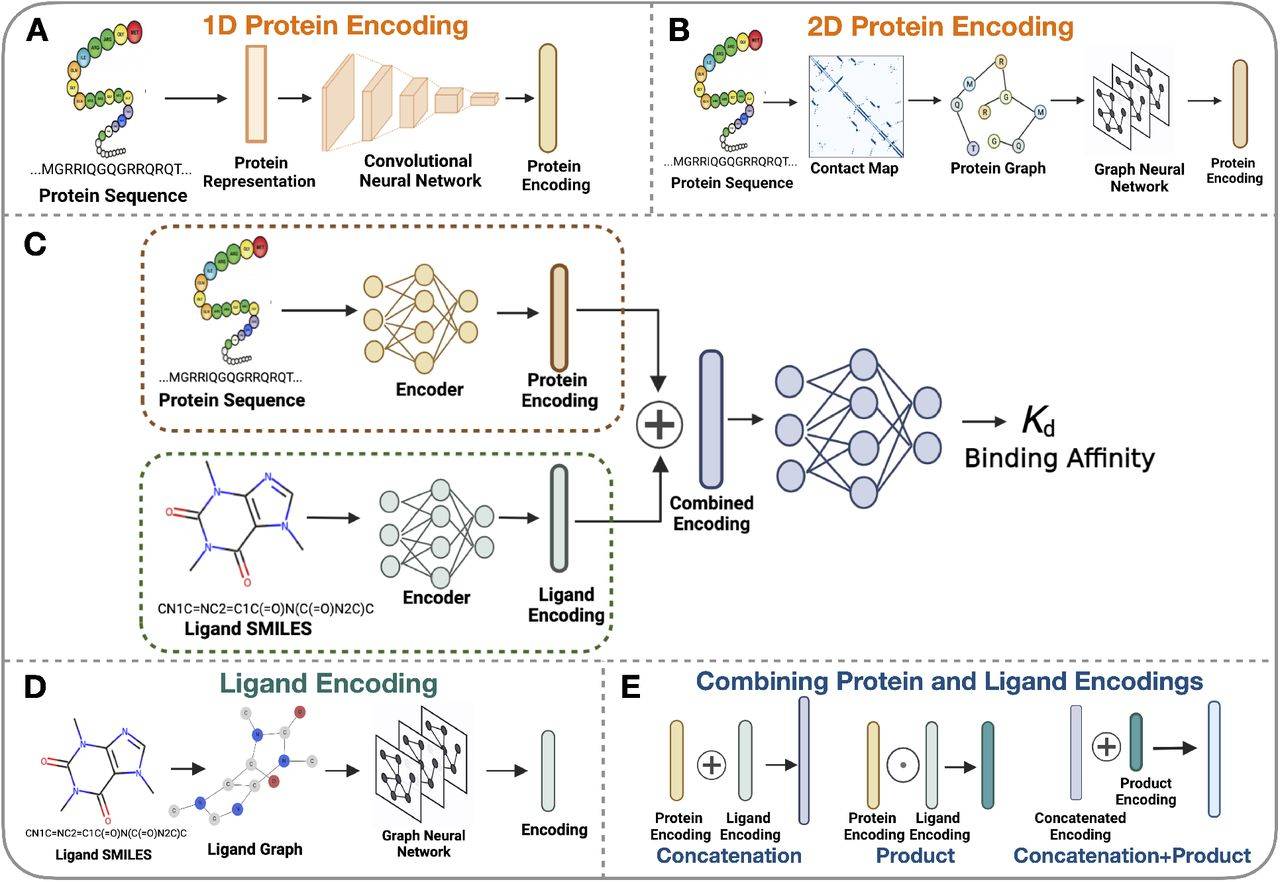
For proteins, they provide sequential information (like the letters of a word) and also patterns of interactions between parts of the protein (like analyzing a map). For ligands, they use the information in their structures. They also use information on how proteins and drugs are combined. They run a lot of experiments to analyze the relationship between the information given to the model and the accuracy with which it predicts the binding affinity.
Methods Employed for Predicting Binding Affinity with Deep Learning
Two types of kinase datasets were utilized in this study, Davis and KIBA. Davis consists of measurements of the strengths of the interactions between kinases and inhibitors, and KIBA combines various sources about how well various compounds interact with specific proteins. The datasets were divided into parts for training and testing the models.
By employing various encoding schemes, the researchers sought to predict these interactions. They adapted algorithms to encode proteins as sequences of numbers and graphs, and ligands as graphs that represent their chemical makeup. For encoding proteins, multiple methods were employed.
One of the methods used a language model trained on a lot of protein sequences. Another one utilized a database to track how proteins interact with other molecules. Then, these encodings were fed as inputs into neural networks. In the case of ligands, their chemical structures were converted into graphs, and the graphs were transformed into numerical encodings.
A complex deep learning model was built that combined these encodings to predict the strength of the interactions between the proteins and ligands. The researchers also tested how the predictions changed when small changes were made in the graphs representing the proteins and the ligands.
At last, statistical measures like Wilcoxon’s signed rank test were employed to evaluate how well the predictions matched the actual data.
An Overview of the Performance of the Model
Comparing PCM Prediction Methods
- The study compared the methods used for predicting protein contact maps (PCMs), or “graphs.”
- Three methods were employed for PCM generation. They were AlphaFold2, ESM-1b, and Pconsc4.
- The highest and lowest accuracy was shown by AlphaFold2 and ESM-1b, respectively.
Impact of Protein Graph Structures on Binding Affinity Prediction
- Protein graphs were used to represent the relationships between different parts of a protein structure.
- Astonishingly, changes in the protein graphs did not have any significant impact on the accuracy of the prediction of binding affinities.
- This indicates that the model does not rely on protein information as much for predicting binding affinities.
Comparing Different Protein Encodings
- The study also compared 1D encodings (sequences) and 2D encodings (graphs) of proteins.
- It was found that just like graphs, the sequences or one-dimensional encoding did not have any substantial impact on the prediction of binding affinities.
Impact of Ligand Encodings
- Ligand encodings were found to have a considerable impact on binding affinity prediction.
- Changing ligand encodings led to significant changes in the model’s performance, with some rendering it highly inaccurate.
Impact of Protein and Ligand Representations
- Ligand representation had a much larger impact on prediction accuracy than protein representation.
- The point randomization method for ligand representation demonstrated the smallest drop in accuracy when changing ligand representation.
Impact of Combining Ligand and Protein Representation
- Multiple methods were employed to combine the representations of proteins and ligands to ascertain whether combining the representations had any impact on the prediction accuracy.
- It was found that combining the representations of proteins and ligands had little impact on prediction accuracy.
Performance of Protein Language Model (ESM-1b) Encodings
- Handcrafted encodings were outperformed by protein language model (ESM-1b) encodings in predicting binding affinities.
- The ESM-1b encodings consistently exhibited high performances across KIBA as well as Davis datasets.
Conclusion
To find out why a machine learning model gives a certain result, it is essential to understand what exactly the model is learning from the information on which it is trained. In this study, the researchers tested a deep learning model in predicting binding affinities between proteins and ligands for enhancing drug discovery. The focus was not only on precision but also on statistical significance. This model places greater emphasis on ligand-related factors as opposed to protein-related factors when predicting binding strength. A constraint existed, specifically, that the study’s sample consisted of only molecules and interactions of interest. The scientists consider their model an initial framework rather than a fully formed solution. Still, they are optimistic that incorporating more diverse data and additional information on molecular interactions in 3D space would overcome the limitations and make the predictions even more robust.
Article Source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Neegar is a consulting scientific content writing intern at CBIRT. She's a final-year student pursuing a B.Tech in Biotechnology at Odisha University of Technology and Research. Neegar's enthusiasm is sparked by the dynamic and interdisciplinary aspects of bioinformatics. She possesses a remarkable ability to elucidate intricate concepts using accessible language. Consequently, she aspires to amalgamate her proficiency in bioinformatics with her passion for writing, aiming to convey pioneering breakthroughs and innovations in the field of bioinformatics in a comprehensible manner to a wide audience.





{kind=link}