
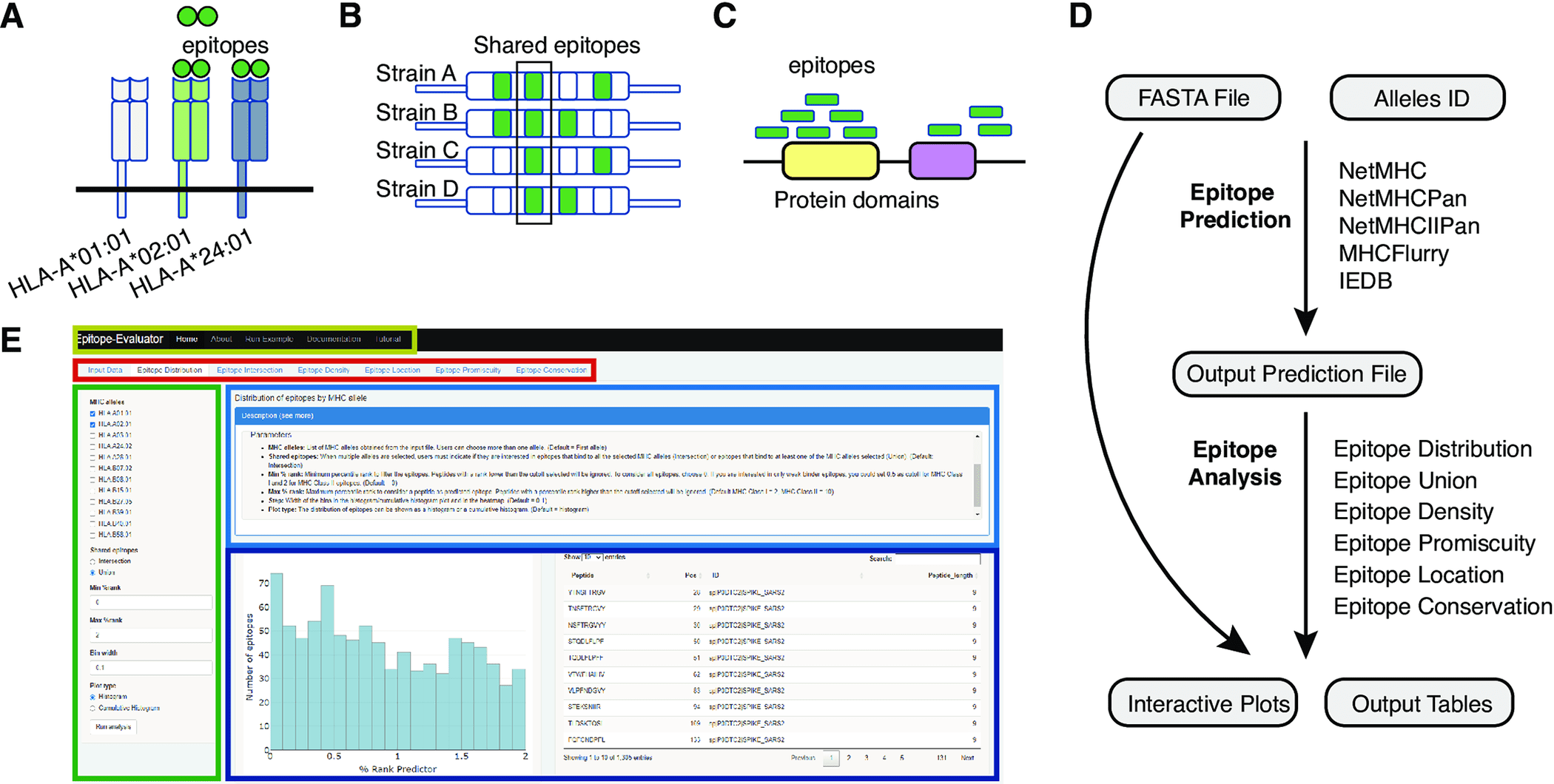
T-cell epitopes are predicted based on amino acid sequences of proteins from different MHC alleles using multiple immunoinformatic tools. Many of these tools produce hundreds of potential peptide candidates, but they are either non-graphical or unfriendly to non-programmers. Epitope-Evaluator is a web application developed in the Shiny/R framework for analyzing predicted T-cell epitopes interactively.
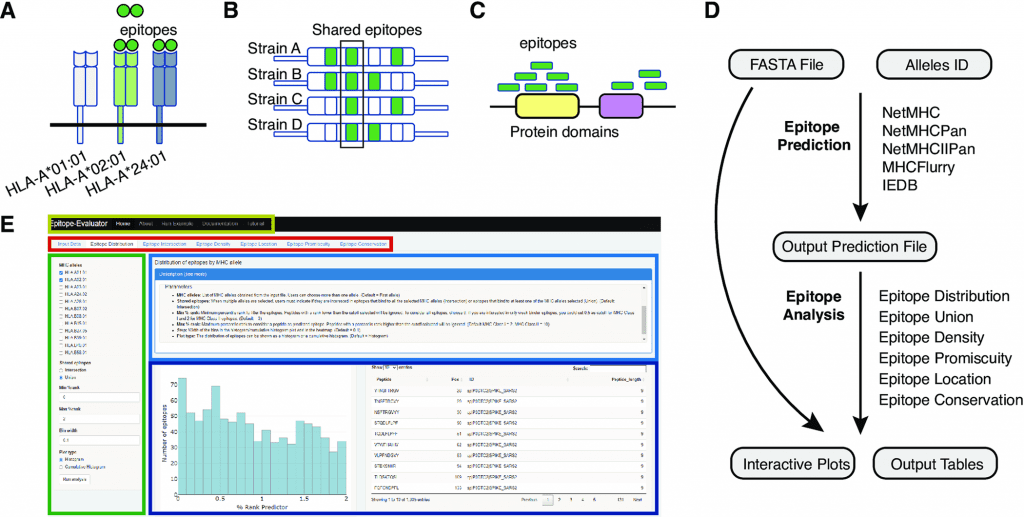
Image Source: https://doi.org/10.1371/journal.pone.0273577
A T-cell epitope is a peptide derived from processed antigens that elicits an adaptive immune response. A CD4+ T-cell recognizes epitopes with a length of 13-17 amino acids on the surface of a major histocompatibility complex (MHC) class II molecule, whereas a CD8+ T-cell recognizes peptides with about nine amino acids on the surface of an MHC class I molecule. In this way, T-cells are able to detect pathogens from cancer cells and abnormal self-antigens from them. Based on cytokine secretion assays such as ELISPOTs or ELISAs, epitope-specific T-cell responses in an input sample can be detected. In addition, epitope-specific T-cells are useful for detecting vaccines and deimmunizing proteins. The development of cancer immunotherapy also draws attention to T-cell epitopes since the number of potential T-cell neoepitopes in a tumor has been proposed as a measure of the effectiveness of checkpoint blockade treatments, as well as the use of tumor-specific epitopes to stimulate T-cell responses to tumors.
It is critical to develop tools for analyzing B-cell epitopes and to integrate MHC Class I T-cell epitopes, MHC Class II T-cell epitopes, and B-cell epitopes in the future. To prioritize epitopes of highly expressed genes, gene expression data for the antigens can also be included. Together, this tool will facilitate the interpretation and analysis of T-cell epitopes by immunologists and experimental scientists.
Epitopes from low-throughput and high-throughput studies are included in the Immune Epitope Database and Analysis (IEDB). IEDB considers three types of assays for identifying T-cell epitopes. The first method involves assays measuring MHC binding in vitro to determine which peptides can stimulate T-cells. In the second assay, MHC ligands are eluted and identified by mass spectrometry. Last but not least, the T-cell response is measured after an epitope is recognized.
It is often necessary to further filter the predicted T-cell epitopes before experimental testing using these predictors. In order to select antigens for screening and selection, epitope features such as predicted binding strength, promiscuity (number of MHC alleles they will bind to), conservation across homologs, and amino acid sequence location are important. Vaccines containing highly conserved epitopes may have the advantage of generating a cross-protective response to the original pathogen as well as to other strains and types. To elicit protective responses in mice, conserved epitopes have been targeted against different influenza A and B subtypes. Recent studies have identified conserved epitopes in Spike that may be targeted to combat emerging SARS-CoV-2 variants.
It is also possible to select vaccine candidates by identifying promiscuous epitopes, as promiscuous epitopes afford broader protection to populations with different MHC genotypes. Different pathogens, such as the hepatitis C virus and Plasmodium falciparum, can be targeted with vaccines containing these promiscuous epitopes. Epitope location within antigens can also provide information on regions with high epitope prevalence, which can be beneficial for subunit vaccine development. Researchers found that Spike protein contains more conformational epitopes than linear epitopes in its RBD region.
It is crucial to understand adaptive immune responses and rational vaccine designs when epitope binding strength, promiscuity, conservation, and location are known. Unfortunately, there is not much software that automates the selection and visualization of epitopes. The EpitopopeViewer Java application allows the identification of immune epitopes within the 3D structure of immunological complexes and antigens in IEDB; however, the application does not show information about epitope conservation or promiscuity, and it is not maintained at the moment. A number of analysis tools are available in the IEDB, including coverage analysis, epitope conservancy, cluster analysis, and mapping mimotopes to antigens, among other things.
Non-programmers are less likely to be able to use these methods since most of them require some level of programming knowledge. As a result of these shortcomings, a user-friendly Shiny app known as “Epitope-Evaluator” was created. It allows non-programmers to analyze T-cell epitopes by identifying conserved or promiscuous epitopes or epitope-enriched protein regions through a graphical interface.
A comprehensive epitope analysis can be performed with the Epitope-Evaluator, which includes six tools and interactive plots. In addition to identifying proteins and regions for the development of peptide-based vaccines, these tools can be used to identify promiscuous and conserved epitopes for the development of multiepitope vaccines and to study how mutations influence the creation of neoepitopes. In order to design a rational vaccine, other aspects associated with the biological function of each protein need to be considered, in addition to epitope binding strength, location, and other sequence-based parameters. Pathogen structure, expression level, and potential toxicity of the proteins are among the factors to consider. Epitope-Evaluator does not consider these parameters since they are case-specific.
A selected set of MHC alleles is analyzed by the Epitope-Evaluator, which also provides information on epitope promiscuity, conservation, and density of epitopes. A protein sequence file and an output prediction file from any predictor are required as inputs for Epitope-Evaluator. Users can download several interactive plots and tables as JPGs and text files by selecting different cutoffs and parameters. The Epitope-Evaluator revealed that HLA-B*40, HLA-B*27:05, and HLA-B*07:02 recognized fewer epitopes from the SARS-CoV-2 proteome than other MHC Class I alleles. Delta, Omicron, and Wuhan Spike variants also shared epitopes, while variant-specific epitopes were identified. In summary, Epitope-Evaluator simplifies experimental epitope selection by providing intuitive tools that allow straightforward interpretation as well as graphical representations.
Article Source: Reference Paper | Epitope-Evaluator Website | Code
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Srishti Sharma is a consulting Scientific Content Writing Intern at CBIRT. She's currently pursuing M. Tech in Biotechnology from Jaypee Institute of Information Technology. Aspiring researcher, passionate and curious about exploring new scientific methods and scientific writing.





{kind=link}