
Researchers from Stockholm University, Sweden, have developed a novel bioinformatics pipeline to predict the structure of large protein molecules, such as the nuclear pore or the 26S proteasome complex, using AlphaFold and Monte Carlo Tree Search.
A protein complex is a group of two or more polypeptide chains associated together. These biological entities play a crucial role in fundamental cellular functions such as transcription, translation, and signal transduction. They also aid in performing other tasks, such as mRNA splicing, protein degradation, and protein folding.
The protein complex map “hu.Map 2.0” is a machine learning framework developed by Drew et al. which combines protein complexes identified from over 15,000 experiments and has shown a total of 4779 multimeric protein complexes. Out of which, only a few are available in the protein data bank (PDB), and out of 3000 eukaryotic protein multimers, only 800 are structurally resolved, thus indicating a gap in the protein structural knowledge.
Modeling protein complexes via homology modeling have enabled scientists to unravel a protein’s function by its structure. There are three major approaches,
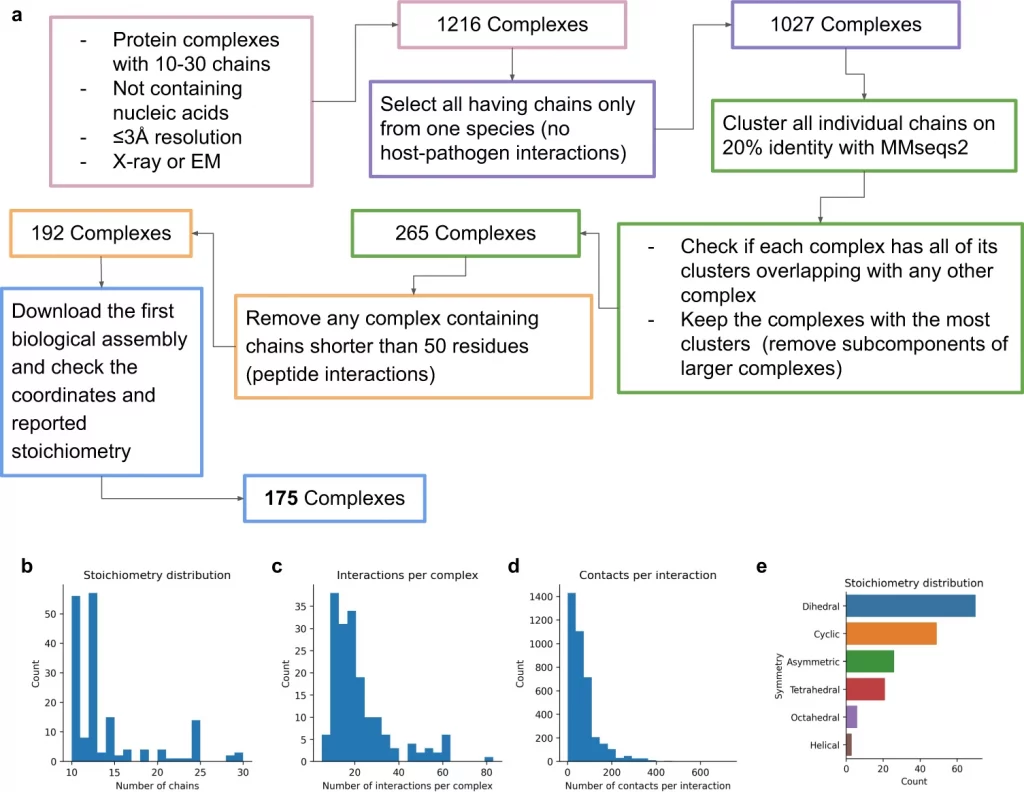
Image Source: https://doi.org/10.1038/s41467-022-33729-4
1) Template-based approach
2) Shape complementarity docking
3) Integrative modeling
Out of which, AlphaFold overthrows the first two approaches. AlphaFold is an AI system developed by DeepMind to predict the 3D structure of any protein, provided its amino acid sequence is known. AlphaFold can help us unravel the structure of dimers and even multimers if a few more docking steps are included in the pipeline. But this methodology cannot help predict the structure of uncommon protein chains. AlphaFold’s predictive power and accuracy decline rapidly for proteins with more than two polypeptide chains. Therefore to overcome this, Bryant et al. have created a graph traversal algorithm that predicts the assembly of protein molecules of up to 10-30 chains in a step-wise fashion and eliminates overlapping interactions.
Findings
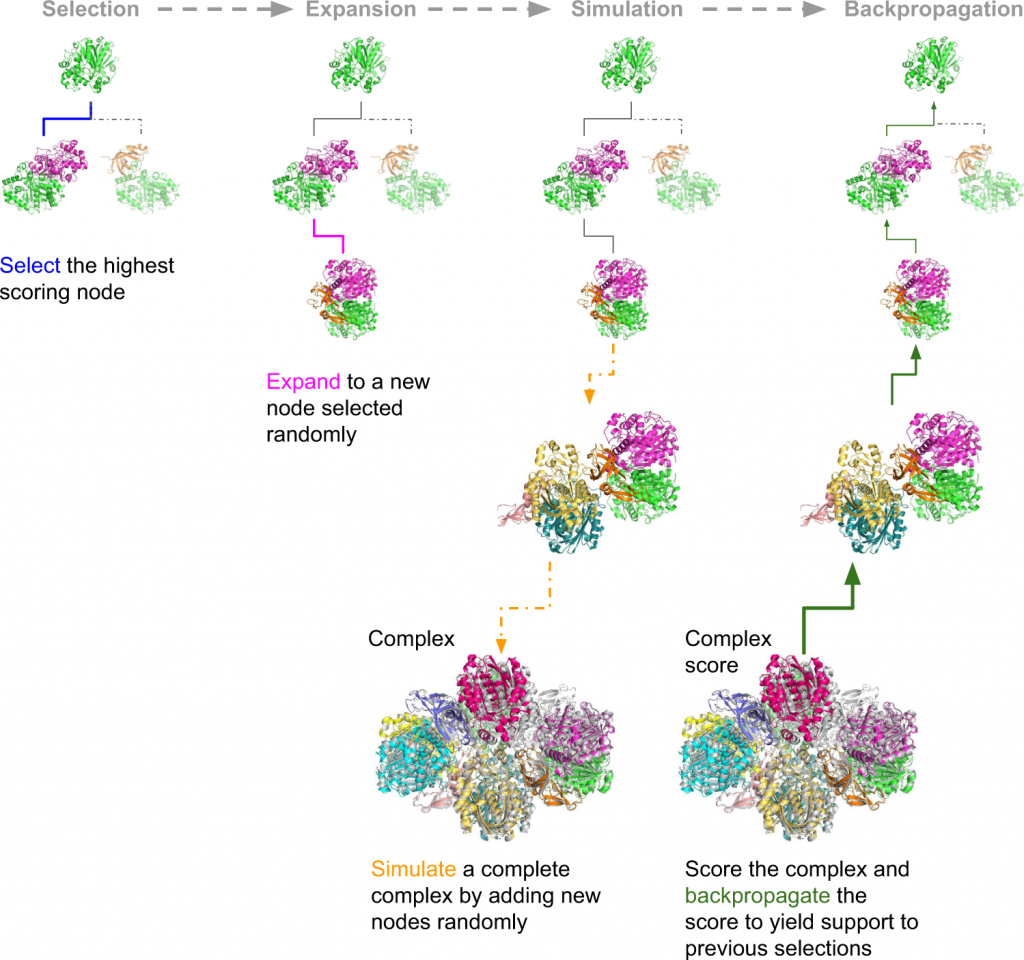
Image Source: https://doi.org/10.1038/s41467-022-33729-4
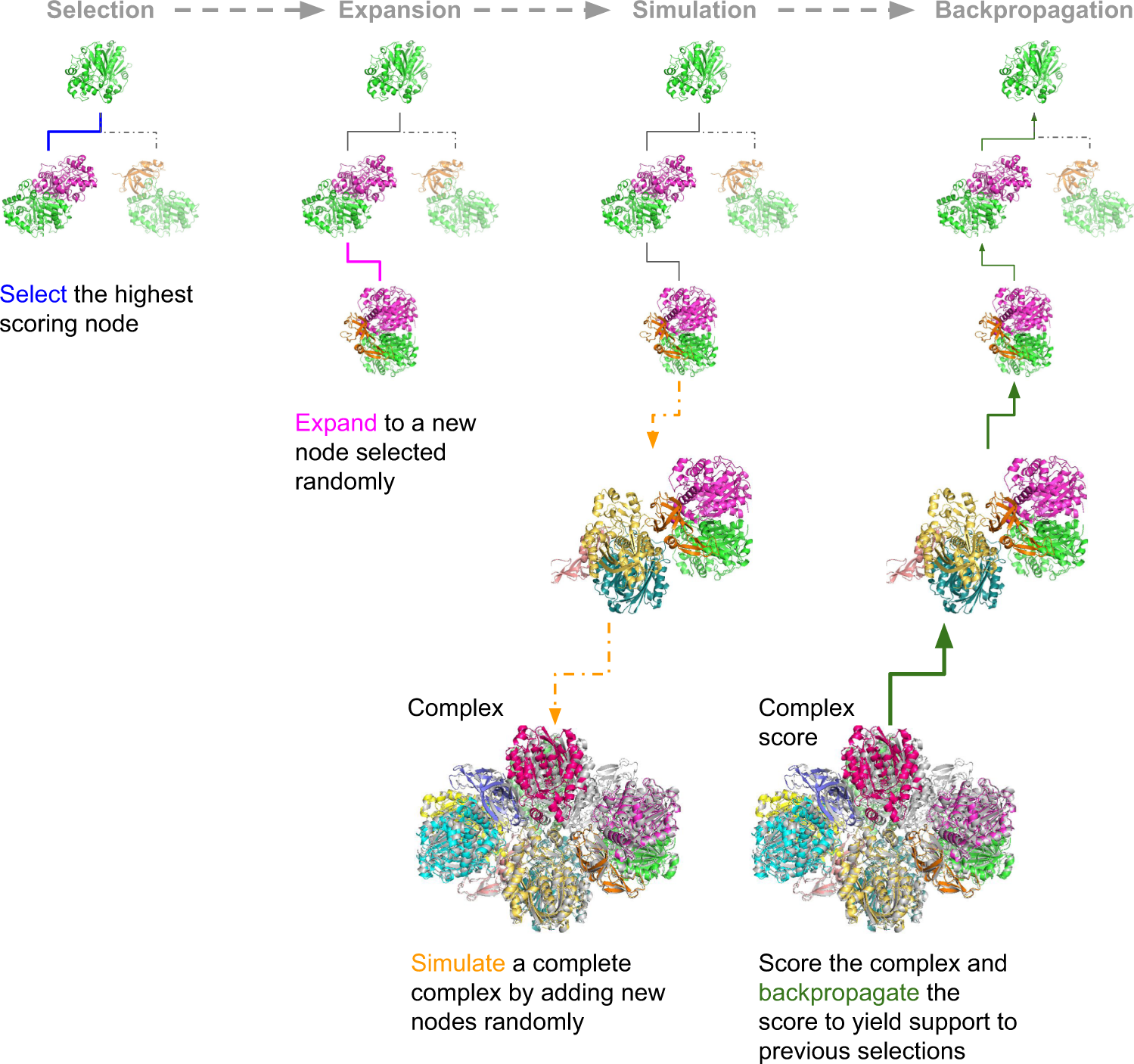
The assembly of protein complexes is predicted using the Monte Carlo Tree Search (MCTS). MCTS is an AI search algorithm that determines the best fit out of all plausible fits. The success rates of the predictions are further examined by AlphaFold Multimer (AFM) or the FoldDock protocol of AlphaFold. The authors’ retrieved structures of multimeric protein complexes from the PDB. For example, the assembly of 6ESQ (Acetoacetyl CoA thiolase/HMG-CoA synthase complex) was predicted using AFM.
With endless possible pathways to assemble, MCTS helps choose the optimal path starting from a random node and expands to form a complete assembly simulation. FoldDock by AF seemingly outperformed AFM in predicting protein trimer. The subcomponents in the protein assembly were predicted with great accuracy using native trimers, suggesting the assembly complexes can be predicted with accuracy, provided the native subcomponents are accurate, indicating the model’s robustness. The symmetry of the complexes makes the prediction more accurate and more effortless. The limit of AF and AFM is approximately 3000 residues, with nearly 42% of the complexes being larger than the abovementioned residues.
The researchers assembled 91 out of a total of 175 complexes consisting of 10–30 chains using MCTS and a median TM-score (Template modeling score between two protein structures) of 0.51. Nearly 30 complexes exhibited high accuracy with a TM-score of approximately 0.8. They also created a scoring function named mpDockQ (multiple-interface predicted DockQ). mpDockQ can help distinguish the fullness and accuracy of the predicted complex.
Limitation Of The Study – Stochiometry is a major limiting factor, as the number of copies of protein present in a complex is often unknown. This limitation could be overcome by experimental verification of different protein assemblies and novel configurations.
Final Thoughts
The study has shown the proposed computational model’s ability to accurately predict large protein complexes with distinct symmetries, done only using the primary sequence and stochiometry. This study opens up promising opportunities to unravel the structure of all cellular protein complexes, large or small.
Article Source: Reference Paper | Code and Data Availability: GitHub | Web version: Colabnotebook
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Shwetha is a consulting scientific content writing intern at CBIRT. She has completed her Master’s in biotechnology at the Indian Institute of Technology, Hyderabad, with nearly two years of research experience in cellular biology and cell signaling. She is passionate about science communication, cancer biology, and everything that strikes her curiosity!





{kind=link}