

A team of researchers at Digital R&D, Sanofi, USA, developed CodonBERT, a transformer-based large language model that is used for performing tasks related to mRNA protein prediction and analysis. It is trained by using codons as its input; 10 million sequences from a diverse range of organisms have been used to facilitate this. It selects the optimal sequences using the codon data it has been pre-trained on. Considering the fact that mRNA vaccines are on the rise, this model proves to be a highly useful asset; a large number of mRNAs can encode the smallest of peptides and proteins, and optimizing such sequences is crucial for effective vaccine design.
An Introduction to mRNA vaccines
mRNA vaccines are recent alternatives to traditional vaccines with numerous advantages associated with them; they can be produced quickly and at a much faster rate than the latter, are cheaper, safer to use, and exhibit high potency. They have been developed and are currently under development for widespread diseases of global health concern, such as influenza, chlamydia, SARS-CoV-2, and cancers such as melanoma and lung cancer.
An mRNA vaccine’s efficacy is determined by the expression levels of the antigenic protein the sequence encodes. High expression levels of antigenic proteins indicate that they are more effective and require a lower dose to be administered to achieve the requisite immune response. This also makes it easier to manufacture these vaccines and reduces their cost of production as well. Expression levels, in general, affect the immunogenicity, efficacy, and potency of mRNA vaccines. The safety and efficacy of these vaccines can be attributed to the fact that the immune response becomes prolonged for a longer period of time, and the lower dose of administration reduces the severity of reactogenicity – the tendency of a vaccine to produce expected side effects observed during clinical trials.
Optimization of Sequences
The number of codon sequences that encode a certain protein is given by the formula 3n, where n is the number of amino acids contained in the length of the protein’s sequence. This can amount to a large number, and viruses and DNA only require about one or two codons to be relevant to any given study. Therefore, it is important to optimize sequences to identify the codons needed.
Conventionally, optimal codons are chosen using a codon bias that is biologically present in the host. This method has several applications in gene therapy, DNA and RNA-based vaccines, and mRNA therapy. The main problem here is that it does not consider some key properties that can be integral for optimization, such as certain structures of RNA (pseudoknots and stem loops) that are important for identifying non-coding RNAs like aptamers or riboswitches. The secondary structures of mRNA have to be accounted for optimization as well, as they affect the stability of mRNA. The replacement of codons with other similar ones can affect the interactions of the protein with nearby structures. Keeping these issues in mind, it is important to note that individual codons are not enough to optimize and generate proteins with high levels of expression.
CodonBERT: The Inside Story
Natural language processing methods use LLMs pre-trained on text present on a large scale, which is fine-tuned afterward. This can be extended to have biological applications as well. Embeddings of nucleotides have been utilized to enable learning tasks by algorithms. However, these alone are not ideal for protein expression prediction, as they only focus on the non-coding regions and isolated nucleotides.
This is where the BERT model is applied to LLMs to understand the mRNA language. It uses a framework consisting of a multi-head attention transformer-based architecture and is used to predict mRNA degradation and protein expression. Both the pre-trained and fine-tuned versions of the model can learn new biological information fed to them and aid in mRNA vaccine design. It has improved current methods of vaccine design as well.
How CodonBERT Works Its Magic
For studying the generalized application of this model, the novel hemagglutinin flu vaccine dataset was used. mRNA candidates that encode for the antigen for influenza hemagglutinin were designed and made to transfect cells. These antigens had variable coding regions as well as fixed regions that remained untranslated. The expression levels exhibited by the proteins were used as the labels for learning tasks given to the model.
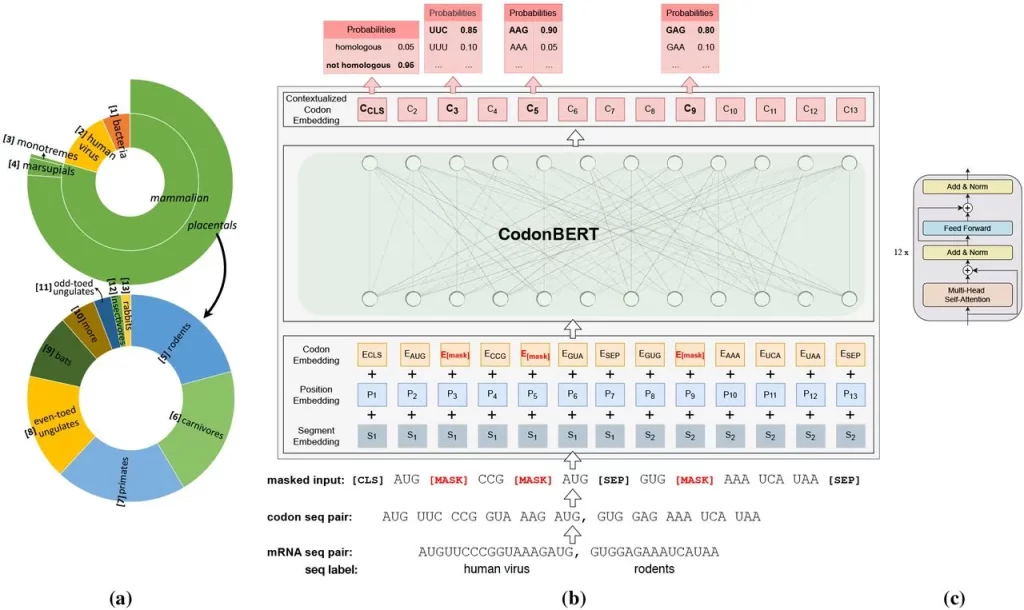
Image Source: https://doi.org/10.1101/2023.09.09.556981
Sequences from all the diverse genome datasets taken into consideration in this study were organized in a hierarchical manner into 13 categories. The input was the coding region, where codons were considered to be ‘tokens,’ whereas the output was the codon placed in a contextual setting. The embeddings had additional layers added to train and fine-tune them to perform prediction tasks.
Two major tasks were carried out for pre-training the model: masked language model learning (MLM) and homologous sequence prediction (HSP). MLM focuses on codons – their interactions, representation, and relations with sequences. HSP is more focused on the sequences of mRNA and the evolutionary relationships between them.
CodonBERT is a self-supervised model that largely relies on MLM and HSP for performing learning tasks. It is capable of both short- and long-range interactions, giving it stability.
Conclusion
CodonBERT is capable of performing two self-supervised tasks, that is, to detect homology and to complete codons, and outperforms prior models in doing so. It can find and record natural selection signals in sequences that support highly expressive and stable mRNA sequences that code for proteins. Based on their respective functions and complexities, this model can also classify genes and organisms. Through the embeddings present in the transformer used, a pseudo-evolutionary tree is produced that aligns with the currently existing knowledge and ideas we hold about evolution as we know it. It has the potential to accelerate the development of mRNA-based vaccines and other innovative vaccines and enhance the production of recombinant therapeutic proteins on an industrial scale.
Story Source: Reference Paper
Learn More:




Swasti is a scientific writing intern at CBIRT with a passion for research and development. She is pursuing BTech in Biotechnology from Vellore Institute of Technology, Vellore. Her interests deeply lie in exploring the rapidly growing and integrated sectors of bioinformatics, cancer informatics, and computational biology, with a special emphasis on cancer biology and immunological studies. She aims to introduce and invest the readers of her articles to the exciting developments bioinformatics has to offer in biological research today.





{kind=link}