
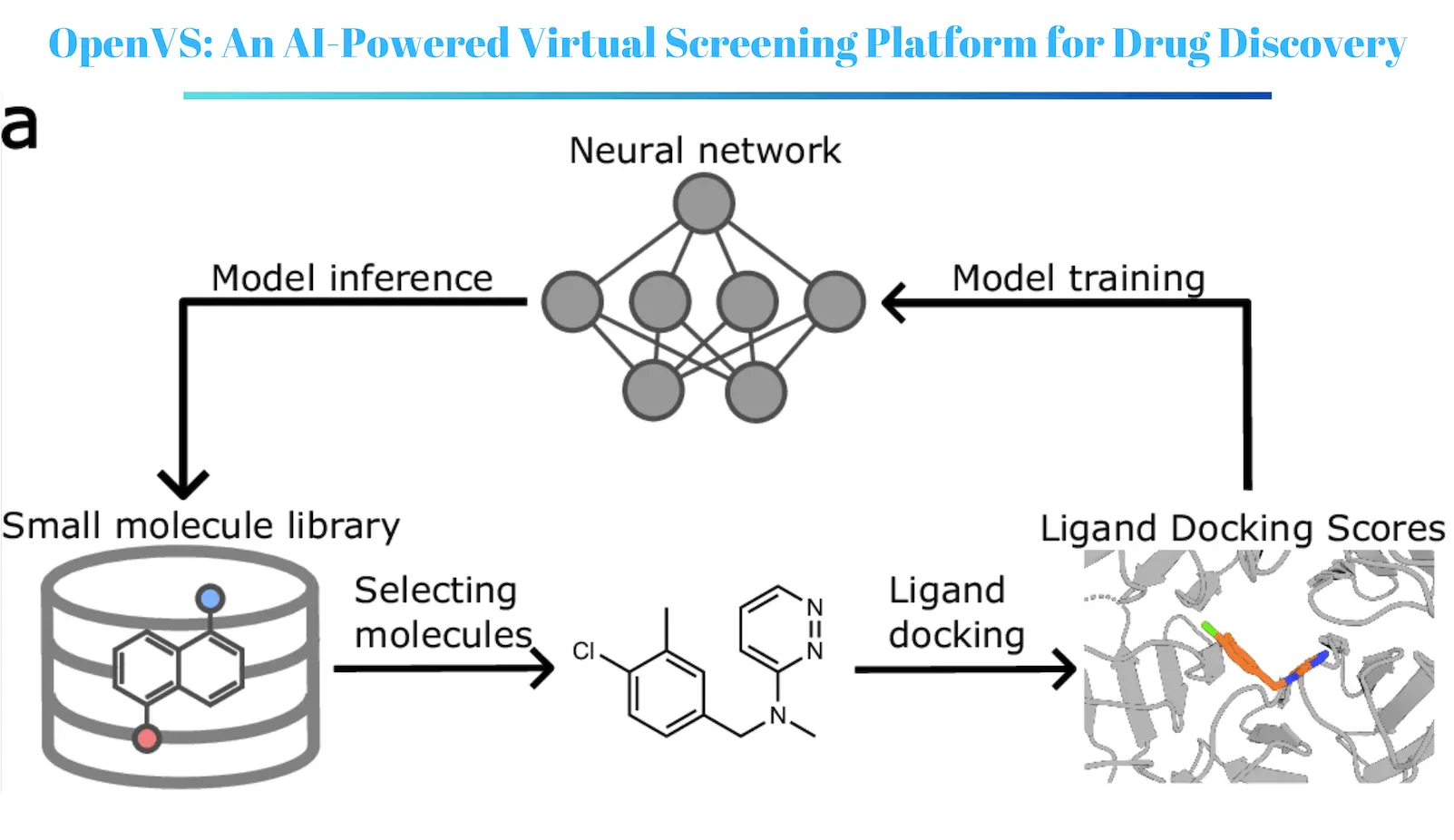
In a recent study published in Nature Communications, researchers at the University of Washington, University of California, San Francisco, and University of Michigan, along with others, revisited the concept of drug discovery by using an innovative AI-augmented virtual screening tool. Named RosettaVS, this technology merges machine learning approaches with a physical model to provide excellent prediction of protein-ligand pairs and pick promising candidate compounds at the end of the process. RosettaVS is incorporated into OpenVS, a new open-source AI-enabled virtual screening platform for drug discovery.
New drug development is an arduous task that takes considerable time and requires a great deal of either in vivo or in vitro experimentation as well as screening a myriad of compounds. The need of the current hour is to find ways to shorten the duration of this process as well as increase the potency of drug production. Seeking to enhance the drug discovery process, a virtual screening process has emerged. Virtual screening is the usage of computer models to estimate the binding capability of small molecules to certain protein targets to screen assay targets.
Understanding Virtual Screening
Virtual screening makes use of computer algorithms to derive small molecules that have the potential to bind to a particular protein target using structural information. Computer simulations of molecular dynamics can replicate the interactions between the small molecule and the protein and predict their binding strengths as well as the possible therapeutic potential of the small drug-like molecule.
Conventional virtual screening techniques rest energy calculations of protein-ligand complexes on a force field potential. More so, these techniques have proven to be very resource-demanding in terms of computation time and cost particularly in vast chemical libraries. To circumvent some of these challenges, people have now pursued dynamic and complex learning systems that can detect and analyze patterns in data, oftentimes referred to as machine learning.
The RosettaVS Platform
The RosettaVS platform designed in this study fully utilizes the advantages of both physics-based potentials and machine learning approaches. A physics-based force field treats protein-ligand interactions more similarly to reality while the screening is done using machine learning to minimize the screening time through pattern recognition of biological data. The platform also takes into account receptor flexibility, – this aspect is very important in many drug targets considered for protein-drug design.
How RosettaVS Works?
The operating procedure of RosettaVS consists of the following steps:
- Protein preparation: The structural constitution of the target protein is done in such a way that the elimination of water and other heteroatoms is performed. The protein is then relaxed to get a lower energy conformation of the protein.
- Ligand preparation: The compounds of the chemical library are here with prepared by the creation of 3D shapes and calculating the physical properties of the models.
- Docking: A force field based on physics is utilized to dock the ligands into the supplied binding pocket of the target protein. This involves modeling the protein-ligand complex and calculating the protein-ligand binding energy.
- Scoring: Poses that have been docked are ranked in terms of binding energy and other parameters such as favorable interactions counted.
- Filtering: In addition, the best poses are ranked and the less biologically active ones are discarded.
- Machine Learning: The model is trained to predict the binding affinity of the ligands based on their features and the docking scores.
- Ranking: The ligands are sorted by the predicted binding affinities in hierarchical order.
RosettaVS incorporates the use of active learning in target identification to reduce costs when dealing with vast chemical collections. This entails training the machine learning model on a small fraction of the library, followed by predicting the binding affinities for the remaining molecules and training the model with those molecules. The highest-scoring molecules are then subjected to docking and evaluation, and the system is updated based on the new data. This continues until a predetermined number of leads have been derived.
Application of RosettaVS to Drug Discovery
Using RosettaVS, the researchers screened billion-membered libraries of compounds against two unrelated targets. One of the targets is KLHDC2, a ubiquitin ligase, and the other is NaV1.7 which is a voltage-gated sodium channel. Compounds with low micromolar affinity were able to be hit with the platform against both targets.
For KLHDC2, the researchers reported the synthesis of seven hit compounds, the hit rate being 14%. One of them, C29, was taken up for further analysis as a potential substrate for KLHDC2 binding. The predicted binding pose of C29 was performed to the test and confirmed by X-ray crystallography.
As for NaV1.7, four hit compounds were identified by the researchers, with a hit rate of 44%. Z8739902234 was one of the four compounds tested and revealed high potency and selectivity for NaV1.7 within inactivated state-dependent assays.
Conclusion
The construction of the RosettaVS platform has contributed to improved performance in several aspects of drug development processes. It can quickly search and find potential drug candidates in massive libraries by uniting two techniques of RosettaVS: computational force fields and neural network-based learning. The drug candidates identified through the platform are expected to be developed in a shorter time than standard manual drug discovery processes would take.
The advancements in drug discovery brought by artificial intelligence are just the start of many applications to come. There is a growing consensus that virtual screening platforms like RosettaVS will be essential in speeding up the identification of novel drugs and their development.
FAQs
RosettaVS is the platform that encompasses drug discovery using virtual drug screening integrated with AI technology. With its unique structural searching method, the software combines the best of the two worlds – the advanced machine learning models with a physics-based force field to model the protein-ligand interactions accurately and in this way screen for the best drug candidates.
RosettaVS functions primarily by computing the binding energies of small molecules that are unambiguously fitted into the binding pocket of a target protein. In this consideration, a docking of highly probable binders of the molecules is first performed to avoid the wastage of computational resources and increase the accuracy of the screening procedure.
Advantages of using RosettaVS include the following:
Precision: It is capable of precisely predicting protein-ligand interactions and quick actions in the search for potent drugs.
Speed: Large libraries of organic molecules can be rapidly analyzed.
Adaptability: It can take into account the receptor’s mobility, which is critical for many drug targets.
Capacity: The tool can in one search, screen billions of libraries of compounds.
Article Source: Reference Paper | RosettaVS is within Rosetta software and is available freely for non-commercial users on GitHub | OpenVS code and scripts are available on Zenodo and GitHub.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}