
Recent advancements in SRT (Spatial Resolved Transcriptomics) technology has allowed in-depth research to measure gene activity in tissue samples and map the activity based on their composition. A collaborative team of researchers from China has introduced a multi-view graph collaborative model, ‘stMVC’ (Spatial Transcriptomics data analysis by Multiple View Collaborative-learning), to analyze SRT data and elucidate tissue heterogeneity to understand these biological compositions better.
Introduction
The golden age data is frequently evolving in different dimensions with the availability of ginormous data. One such dimension is biological information, living beings have been associated with huge accumulated data derived from understanding genomic, proteomic, metabolomic, and transcriptomic levels. Similarly, technology to profile gene expression with context tissues and their cellular organization has evolved rapidly. Spatially resolved transcriptomics (SRT) which includes image-based and sequencing-based technology to profile tissue insights based on tissue histology, spatial location, and tissue polymorphism, is one such advancement.
Due to the immense potential of SRT technology and data, it is highly regarded to unfold developmental and disease biology. Despite the efforts, the SRT data analysis for discovery in biomedical sciences remains difficult due to low throughput, little sensitivity of tools, and distorted data. Some of the introduced computational approaches to analyze the SRT data include Giotto, which utilizes a processing strategy equivalent to single-cell RNA sequencing, SpaGCN using graph convolutional network to identify spatial domains and associated genes, DR-SC and SC-MEB utilizes the hidden Markov random field modeling, STAGATE integrates both gene expression and spatial domains assisted by graph attention auto-encoder framework.
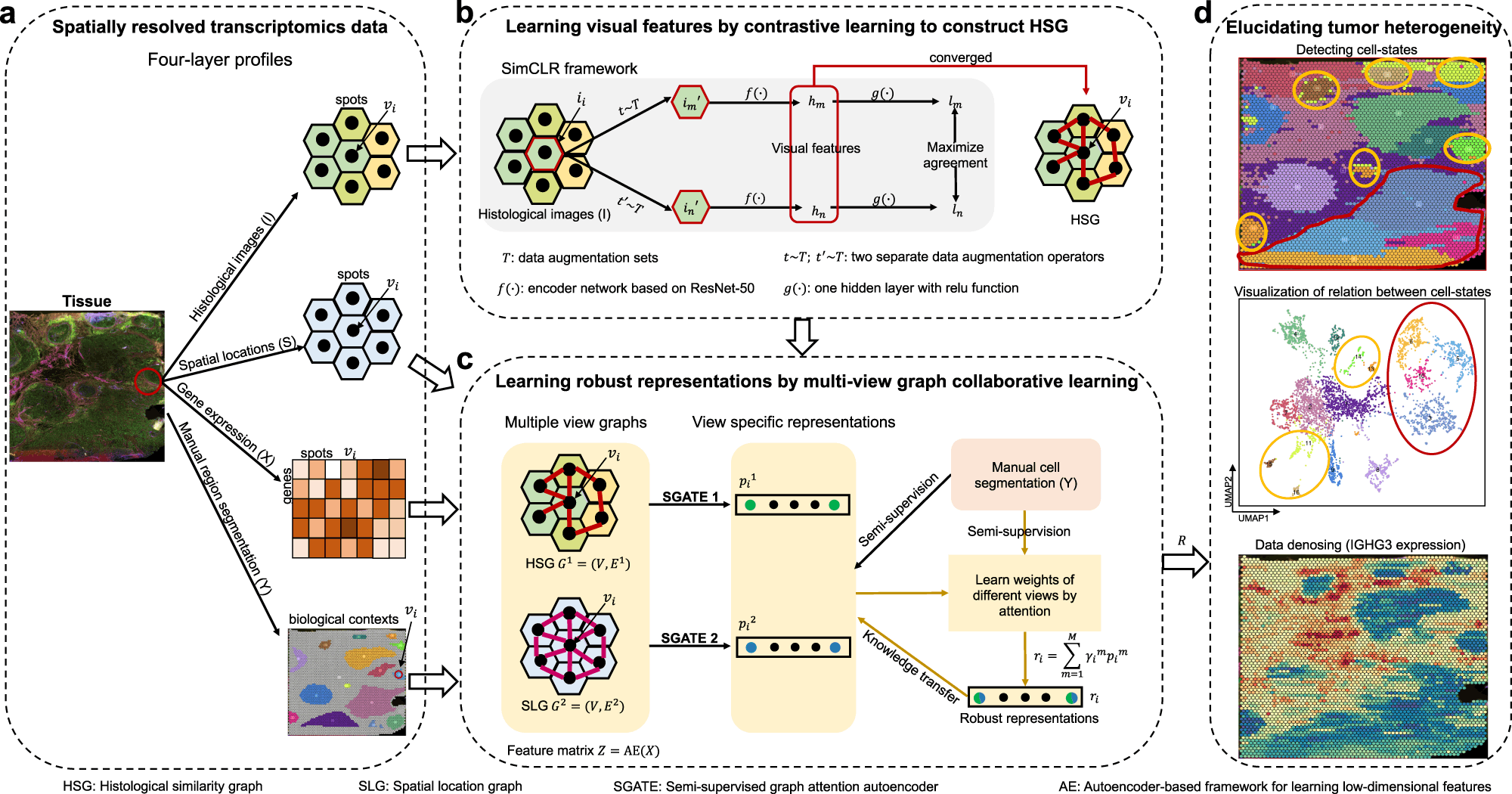
While each model has provided a benchmark in analyzing SRT data, these lack features in extracting data in terms of biological contexts, such as tissue histology and the organization of cells to decipher developmental biology. Collaborative research from scientists from the Chinese Academy of Sciences, Donghua University, and Nanjing Medical University constructed a multi-view graph collaborative model stMVC that integrates tissue histology, spatial location, and tissue heterogeneity while analyzing SRT data.
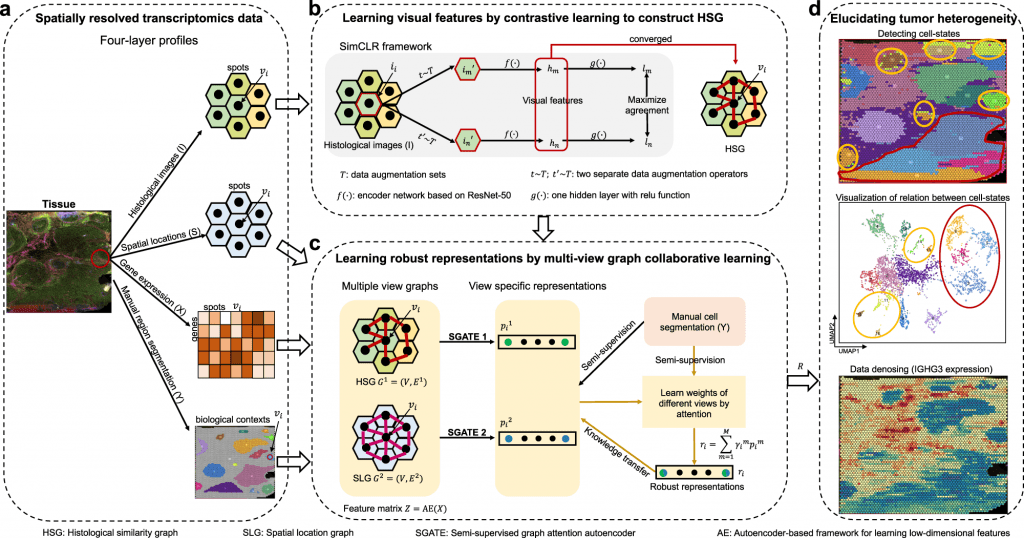
Image Source: https://doi.org/10.1038/s41467-022-33619-9
The stMVC model allows visualization of tissue features obtained by the ResNet-50 model framework. Researchers have emphasized the advantages of the model through comparative analysis providing its efficiency to characterize tissue polymorphism, single-view graph representation by SGATE (semi-supervised graph attention encoder), evaluate multi-view graphs, and low-dimensional representation encrypted from data of gene expression.
Scientists processed 12 slices of the human dorsolateral prefrontal cortex (DLPFC) dataset to reveal the trajectory relationship between the different spatial domains. For comparative visualization and clustering analysis, other methods such as Giotto, DR-SC, and STAGATE were applied. Compared to other methods, stMVC outperformed in terms of clustering accuracy and detecting accurate structure displaying the advantages of attention-based multi-view integration over mean-based strategy. For spatial domains, the model displayed chronological order reflecting relationship trajectory elucidating the complexity of these domains.
Considering the heterogeneity displayed by cancerous cells in tumors, Zuo et al. and team analyzed ovarian cancer and breast cancer data publicly published by 10X genomics. A total of 18 regions for ovarian cancer and 16 for breast cancer were annotated independently. The regions were classified into different stages of cancer: invasive carcinoma, carcinoma in situ, and benign hyperplasia. stMVC model detected more spatial domains with enriched cancer regions as compared to other computational models. The model effectively detects five regions of breast cancer, while others detected three regions.
The model clearly stated advantages in ovarian cancer, such as domains 11 and 12 enriched with different cell populations, which were limited by single-cell RNA sequencing. stMVC mapped influenced the functioning of four domains which were influenced by infiltrating immune cells as domain 10 showed infiltration by CD8+ T cells, and domain 12 showed overexpression of M2 macrophage resulting in secretion of TGF-β. The method was also capable of identifying normal healthy calls from the pool of cancerous cells based on histological features such as cell size and shape.
Supportive from the earlier findings, the team also demonstrated the stMVC model on the mouse primary visual cortex (VI) 1020 gene dataset obtained from the STARmap platform. In order to detect layer-specific inhibitory neurons, the RNA clusters per cell were predicted using ClusterMap and annotated seven different layers. The model revealed how each cortex is assigned by different feature embeddings with both excitatory and inhibitory neurons spread throughout the canonical layers, followed by the detection of layer-specific inhibitory neurons, i.e., Sst+ and Pvalb+, consistent with earlier studies.
Final Thoughts
The research team proposed a new model to analyze the SRT data effectively and apply the findings to understand the tumor heterogeneity and illustrate functional brain datasets. Demonstrating models on real-life samples of breast and ovarian cancer helped detect cell states, metastasis, and cell infiltration. stMVC provided better performance in terms of spatial trajectory, clustering, and also reducing noise in the analyzed datasets. The future implications can be drawn to show functional regions and layer-specific cell states in the brain, similarly, it can provide potential diagnostic applications in detecting tumors and metastasis. Some of the shortcomings of manual annotations for region segmentation within the tumor dataset and exploration using deep learning models can be sufficiently addressed for the efficient automatic structure of the model.
Article Source: Reference Paper | Code Availability: Zenodo | stMVC Tool: GitHub
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Mahi Sharma is a consulting Content Writing Intern at the Centre of Bioinformatics Research and Technology (CBIRT). She is a postgraduate with a Master's in Immunology degree from Amity University, Noida. She has interned at CSIR-Institute of Microbial Technology, working on human and environmental microbiomes. She actively promotes research on microcosmos and science communication through her personal blog.





{kind=link}