
Understanding cellular responses to genetic changes holds vital importance in various biomedical contexts, such as uncovering cancer-related genetic interactions and advancing regenerative medicine. However, the vast number of potential multigene alterations poses a challenge for practical experimentation. In this regard, a novel method called the graph-enhanced gene activation and repression simulator (GEARS) has been introduced by scientists from Stanford University. GEARS combines deep learning with a gene relationship knowledge graph to forecast transcriptional reactions to single and multiple gene perturbations, utilizing single-cell RNA-sequencing data from perturbation screens. Impressively, GEARS can even predict outcomes for gene combinations that have never been experimentally perturbed. Compared to existing techniques, GEARS demonstrated 40% higher precision in predicting distinct genetic interaction types and outperformed prior methods in identifying strong interactions. Ultimately, GEARS has the potential to anticipate the diverse effects of multigene alterations, offering valuable guidance for the design of perturbation-based experiments.
The Importance of Effectively Predicting Genetic Perturbations
Understanding how cells respond to genetic changes is crucial for unraveling their functions, and these insights extend from maintaining cellular identity to reversing disease traits through gene expression modulation. Such revelations hold significant implications for biomedical research, particularly in tailoring personalized therapies. For instance, validating drug targets using genetic perturbation aids successful clinical trials, while identifying synergistic gene pairs enhances combination therapies. As complex cellular behaviors often stem from interactions among small gene sets, recognizing these interactions could refine precise cell engineering. While recent strides enable swift experimental sampling of perturbation outcomes, computational methods are essential for prioritizing experiments due to the overwhelming possibilities of multigene combinations.
However, prevailing computational approaches for predicting perturbational outcomes have limitations. Mainly, single-gene perturbation predictions rely on deducing transcriptional links within gene regulatory networks, constrained by challenges in accurate network inference from gene expression data or incomplete databases. Existing models built on such networks linearly combine individual perturbation effects, failing to foresee non-additive multigene effects like synergy. Some recent approaches employ deep neural networks trained on large perturbational data to sidestep network inference and predict outcomes directly from genetic relationships. Nonetheless, these methods still demand experimental perturbation of each gene in a combination before predicting its combined effect.
GEARS merges deep learning with a gene relationship knowledge graph to simulate genetic perturbation effects. Infused with biological knowledge, GEARS predicts outcomes for single genes or gene combinations even lacking prior perturbation data. GEARS surpassed existing methods in predicting outcomes for both single and dual gene perturbations from diverse datasets, detecting various genetic interaction subtypes, and extrapolating to new perturbational territories by predicting unprecedented phenotypes. This capacity positions GEARS to influence the planning of forthcoming perturbational experiments significantly.
How GEARS Predicts Perturbations
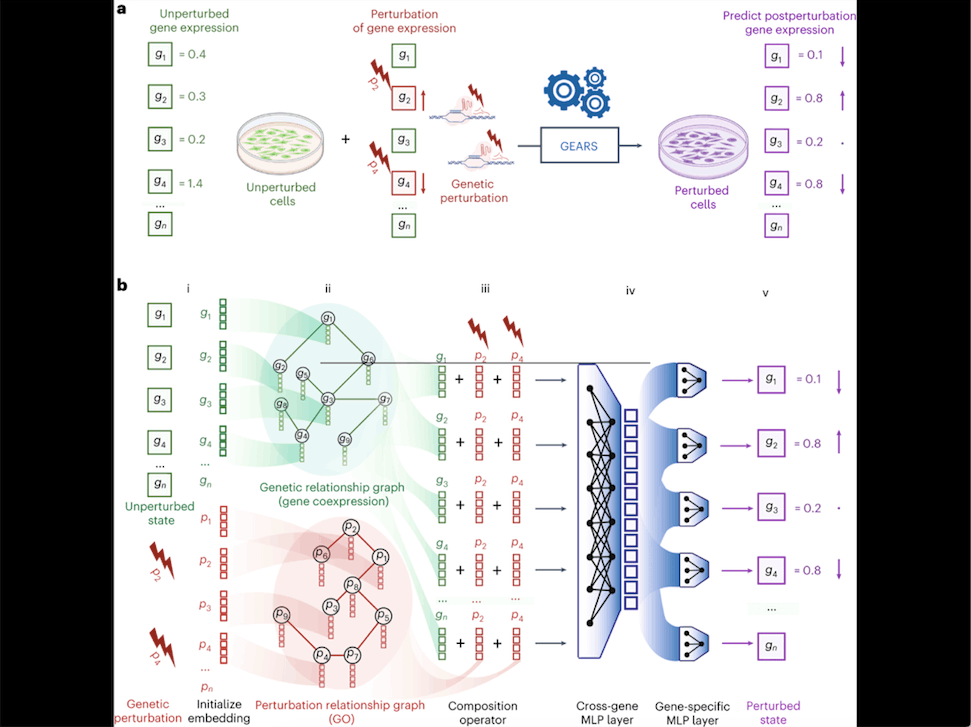
1. Perturbation and Prediction: GEARS predicts how the expression of genes changes when a set of genes is perturbed. Given the baseline gene expression data of individual cells and the perturbation set applied to those cells, GEARS generates predictions about how the cells’ gene expression will be altered.
2. Embeddings: GEARS uses “embeddings” to represent genes and their perturbations. Embeddings are multidimensional numerical vectors that capture meaningful characteristics of genes and perturbations. Each gene has its own embedding that is trained to capture important traits of that gene.
3. Multidimensional Components: The gene embeddings are split into two multidimensional components. This allows GEARS to capture genes’ unique variability and response to perturbations more effectively.
4. Prediction Process: The gene embeddings and perturbation embeddings are combined in sequence to predict the post-perturbation state of the gene. This prediction takes into account the “cross-gene” embedding vector, which represents overall transcriptome-wide information for each cell.
5. Incorporating Prior Knowledge: GEARS leverages existing biological knowledge to enhance its predictions. It incorporates gene-gene relationships from a coexpression knowledge graph and uses Gene Ontology-derived knowledge to understand the impact of gene perturbations on other genes.
6. Graph Neural Networks (GNNs): GEARS employs a graph neural network (GNN) architecture to leverage the information from knowledge graphs. GNNs are specialized neural networks designed to work with graph-structured data, such as networks of interconnected genes and their relationships.
7. Biological Intuitions: GEARS relies on two biological intuitions: genes with similar expression patterns are likely to respond similarly to perturbations, and genes involved in similar pathways are likely to affect the expression of related genes after perturbation.
The Many “Achievements” of GEARS
- Single gene perturbation prediction: Evaluating the model on genes that were excluded from training, GEARS demonstrated its efficacy on two datasets comprising thousands of perturbations and over 170,000 cells. Comparing GEARS with baseline models, it exhibited substantial superiority, yielding 30-50% improvements in mean squared error and over two times better Pearson correlation values. Importantly, GEARS accurately captured the direction of gene expression changes, highlighting its enhanced understanding of regulatory relationships. These results remained consistent across various datasets, including genome-wide perturbation screens, and showcased its scalability compared to traditional gene regulatory network-based methods. Impressively, GEARS extended beyond transcription-level prediction, identifying gene clusters with similar responses to perturbations, even for genes unseen during training.
- Multigene Perturbation Prediction: GEARS focuses on predicting transcriptional outcomes for multigene perturbation sets. Evaluated on a Perturb-seq dataset, it addressed two-gene perturbations. Different scenarios were considered: both genes seen, one of two unseen, and both unseen during training. GEARS consistently improved performance by over 30% across cases, even 53% when both perturbed genes were unseen. Individual gene analysis showed accurate trend and magnitude prediction, even for genes unseen during training. Incorporating knowledge graphs was crucial but constrained predictions for less-connected genes. GEARS used Bayesian formulation to output an uncertainty metric inversely correlated with model performance, overcoming these challenges. It showed 50% greater enrichment in significant differentially expressed genes compared to baselines.
- Non-additive combinatorial perturbation prediction: In the context of two-gene perturbations, a simple addition of individual perturbational effects may not accurately estimate combinatorial effects due to non-additive genetic interactions. GEARS identified five interaction types and demonstrated a stronger correlation between its predicted genetic interaction scores and true scores compared to existing methods. The model recommended gene pairs likely to exhibit strong interactions, resulting in over 40% improvement in Precision@10 and a twofold accuracy increase for the top ten strongest interactions. Additional validation confirmed GEARS’ effectiveness across genetic interaction subtypes. Even with only one gene’s perturbation history, GEARS successfully detected synergistic and suppressive interactions. At the gene level, it captured genetic interaction effects over 40% better than other methods across various subtypes.
- Novel phenotype prediction: GEARS was employed to predict outcomes for pairwise combinatorial perturbations involving 102 genes from the Norman et al. dataset. By training on both one-gene and two-gene perturbation expression profiles, GEARS successfully captured distinct phenotypic clusters and identified novel phenotypes not observed in training data. Among these, a cluster displaying a high expression of erythroid markers emerged. To confirm the biological relevance of this novel phenotype, it was compared with proerythroblast data from the Tabula Sapiens cell atlas. Despite lacking experimental validation, its discovery showcased GEARS’ potential to uncover unobserved post-perturbation phenotypes. Robustness was validated by excluding similar outcomes during training.
- Genetic interaction mapping: Extending their analysis, the researchers predicted genetic interactions for all possible pairwise combinations of 102 genes using CRISPRa-based gene activation. Leveraging post-perturbation gene expression predictions for the 5,151 combinatorial perturbations, they constructed a comprehensive genetic interaction map encompassing five interaction subtypes. The map revealed a diverse landscape of interactions, especially between functionally related genes. The uniqueness lay in its broader coverage compared to traditional maps, which predominantly focus on synergistic interactions. Validating some predictions with cell fitness data, GEARS demonstrated comparable performance to real Perturb-seq experiments in capturing strong interaction effects. This boosted confidence in the biological significance of captured interactions, even without full experimental validation. Additionally, GEARS directly predicting cell fitness displayed robust performance.
Conclusion
Recent advancements in high-throughput perturbational screens have improved gene targeting precision and data scale. While CRISPR-based screens are gaining traction in drug discovery, GEARS complements such experiments by inferring a wider range of multigene perturbation outcomes from the same data. It aids in screen design by suggesting perturbations that optimize information gain and reduce costs. Reliable predictions require consistent cell type and combinatorial perturbation training. GEARS detects emergent gene interactions, facilitating innovative cell engineering for diverse applications, from cancer treatment to cell reprogramming. This model not only aids in discovering new therapies but also shapes the future of cell- and gene-based treatments.
Article Source: Reference Paper | Code to run GEARS is available at GitHub
Learn More:




Neegar is a consulting scientific content writing intern at CBIRT. She's a final-year student pursuing a B.Tech in Biotechnology at Odisha University of Technology and Research. Neegar's enthusiasm is sparked by the dynamic and interdisciplinary aspects of bioinformatics. She possesses a remarkable ability to elucidate intricate concepts using accessible language. Consequently, she aspires to amalgamate her proficiency in bioinformatics with her passion for writing, aiming to convey pioneering breakthroughs and innovations in the field of bioinformatics in a comprehensible manner to a wide audience.











{kind=link}