
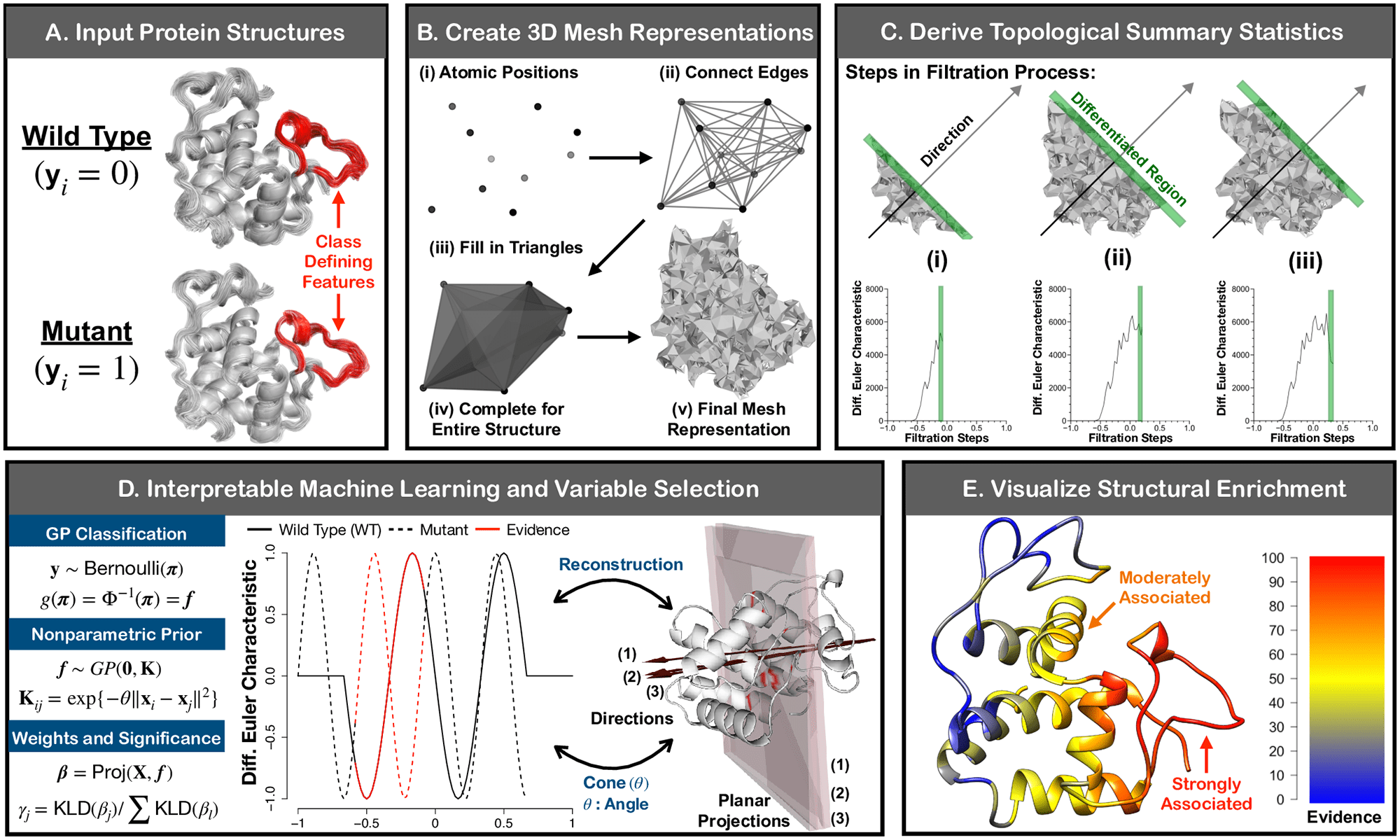
A group of scientists jointly proposed SINATRA Pro, a computational system for the extraction of key structural features between two sets of proteins. SINATRA Pro robustly outperformed standard methods in pinpointing the physical locations of both static and dynamic signatures across different kinds of protein ensembles.
Image Source: A topological data analytic approach for discovering biophysical signatures in protein dynamics
Recognizing structural differences among proteins can be a non-trivial task. While differentiating ensembles of protein structures obtained from molecular dynamics simulations, biologically important features can be easily overshadowed by spurious fluctuations.
In this study, the scientists present SINATRA Pro, a computational pipeline intended to robustly identify topological contrasts between two sets of protein structures.
Algorithmically, SINATRA Pro works by first taking in the 3D atomic coordinates for every protein snapshot and summarizing them as indicated by their underlying topology.
Statistically critical topological features are then projected back onto a user-selected representative protein structure, along these lines facilitating the visual identification of biophysical signatures of various protein ensembles.
The scientists assessed the capability of SINATRA Pro to detect minute conformational changes in five independent protein frameworks of differing complexities.
In all the test cases, SINATRA Pro identified the known structural features that have been validated by past experimental and computational studies, as well as novel features that are likewise prone to be biologically relevant, as indicated by the literature.
These outcomes feature SINATRA Pro as a promising technique for working with the non-trivial task of pattern recognition in trajectories resulting from molecular dynamics simulations, with substantially increased resolution.
Highlighting the Importance of Accurate Characterization of Protein Conformational Dynamics
Recognizing structural features related to macromolecular dynamics is vital to how we might interpret the underlying physical behavior of proteins and their more extensive effect on biology and health.
The structural and dynamical properties of proteins frequently serve as signatures of their functions and activities. Subtle topological changes in protein conformation can prompt dynamic changes in biological function, along these lines featuring the significance of having the option to characterize protein conformational dynamics precisely.
Traditionally, the structural dynamics of proteins have been modeled by the utilization of molecular dynamics (MD) simulations, which work by sampling structural ensembles from conformational landscapes.
In infinite timescales, such structural ensembles are supposed to represent all physical states to such an extent that their ensemble-averaged observables converge to actual physical values and are accordingly physically meaningful.
While MD simulations have given key insights of knowledge into the atomistic motions that underpin numerous protein functions, biologically-relevant structural changes can be overshadowed by spurious statistical noise brought about by the thermal fluctuations that naturally emerge throughout these simulations.
Analysis of Data from MD Simulations and Construction of Functional Correspondences
The information from MD simulations is frequently analyzed in a rigorously goal-dependent manner by the utilization of computational techniques that quantify and assess explicit protein characteristics. For instance, geometric changes that emerge because of ligand binding, point mutations, or post-translational modifications are usually inferred by investigating the root mean square fluctuations (RMSF) of atomic positions or the per-domain radius of gyration concerning a reference structure.
Unfortunately, these standard methodologies are less powerful when the relevant changes in protein structure are overshadowed by fluctuations irrelevant to the biological process of interest.
As of late, more modern techniques are expected to overcome these difficulties by exploiting correspondences between the atomic positions on any two given proteins. For instance, per-residue distance functions or contact maps can be calculated on each frame of a trajectory for clustering or principal component analyses (PCA), which project complex conformities onto a lower-dimensional space for simplicity of comparison.
In any case, the disadvantage to these techniques is that they require diffeomorphisms between structures (i.e., the map from protein A to protein B should be differentiable).
There are numerous scenarios in protein dynamics where no such transformation is guaranteed because atomic features can be acquired or lost during the evolution of the system.
For sure, there are 3D shape algorithms that construct more general “functional” correspondences and can be applied evenly across shapes with varying topology; notwithstanding, past work has shown that the performance of these algorithms drops significantly when the expected functionally mapping input is even marginally misspecified.
The SINATRA Pro
In this work, the scientists present SINATRA Pro: a topological data analytic pipeline for recognizing biologically relevant structural contrasts between two protein structural ensembles without the requirement for explicit contact maps or atomic correspondences.
Their algorithm is an augmentation of a past framework, SINATRA, which was broadly introduced to perform variable selection on physical features that best portray the variation between two groups of static 3D shapes.
By the utilization of a tool from integral geometry and differential topology called the Euler characteristic (EC) transform, SINATRA was displayed to have the ability to identify known morphological perturbations in controlled simulations and robustly recognize anatomical aberrations in mandibular molars associated within four distinct suborders of primates.
SINATRA Pro is defined as an adaptation of the SINATRA framework for protein dynamics. Here, the scientists developed a simplicial complex construction step to explicitly model 3D geometric and topological relationships between atomic positions on protein structures.
The researchers likewise used another set of statistical parameters, which they calibrated for complex protein systems.
SINATRA Pro: Abilities and Promises
In this study, the scientists demonstrated the SINATRA Pro’s capability to recognize critical structural and dynamical features in a hierarchical order of proteins with progressively challenging features to resolve statistically.
The five proteins considered, TEM β-lactamase, the Abelson Kinase (Abl1), the HIV-1 protease, Elongation Factor Thermo Unstable (EF-Tu), and Importin-β, go through structural changes in response to a wide scope of well-studied biological phenomena, including mutations, interactions with partners, and small molecule binding.
The scientists demonstrated that SINATRA Pro outperformed standard analytic techniques, including RMSF and PCA, for reliably pinpointing physical locations of biologically relevant conformational changes.
In general, the researchers observed that SINATRA Pro holds incredible promise for extricating topological differences between two sets of protein structures from meaningless statistical noise.
The Endpoint
There are numerous expected extensions to the SINATRA Pro pipeline. In the first place, in its current structure, SINATRA Pro treats all atomic features as being similarly important a priori to the phenotype of interest.
One especially intriguing extension of the technique would be to up, or down-weight the contributions of various kinds of atomic features (e.g., carbons, hydrogens, or oxygens) or residues (e.g., serine versus arginine) to more precisely represent the topology of explicit inter-atomic connections like hydrogen and covalent bonds.
Practically speaking, this would require making such annotations and deriving topological summary statistics of protein structures in light of a weighted Euler characteristic transform.
Another natural extension would have been to apply the SINATRA Pro pipeline to different data types used to study variation in 3D protein structures like cryogenic electron microscopy (cryo-EM), nuclear magnetic resonance (NMR) ensembles, and X-ray crystallography (i.e., electron density) information.
Past work has proactively shown that topological characteristics computed on tumors from magnetic resonance images (MRIs) can potentially be strong predictors of survival times for patients with glioblastoma multiforme (GBM) and other cancer subtypes; in any case, it has additionally been noticed that the efficacy of current topological summaries diminishes when heterogeneity between two phenotypic classes is driven by minute differences. For instance, cryo-EM images can look quite similar in any event for two proteins harboring different mutations.
The improved, superior ability of SINATRA Pro to capture inter-class variation is driven by local fluctuations in shape morphology, so it would be fascinating to check whether the scientists’ proposed pipeline could offer more resolved insights for these sorts of applications.
Article Source: A topological data analytic approach for discovering biophysical signatures in protein dynamicsTang WS, da Silva GM, Kirveslahti H, Skeens E, Feng B, et al. (2022) A topological data analytic approach for discovering biophysical signatures in protein dynamics. PLOS Computational Biology 18(5): e1010045. https://doi.org/10.1371/journal.pcbi.1010045
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.

, a Leap Forward in Artificial Life Discovery")






{kind=link}