
A newly developed AI model called popEVE shows promising results for accelerating the diagnosis of rare genetic disorders by predicting which mutations in a patient’s genome are most likely to cause severe disease. The model was created by researchers at the Harvard Medical School, USA, and deployed on genome data from over 31,000 patients with rare developmental disorders.
Genetic testing has become an invaluable tool for diagnosing rare diseases. However, even when a patient’s entire genome is sequenced, interpreting the results can be extremely challenging. Each person has around 4-5 million genetic differences compared to the reference human genome. The vast majority of these variants are benign, while only one or two may actually be causing the disease. This creates a major bottleneck in connecting patients’ symptoms to an underlying genetic cause.
Computational prediction methods can help prioritize candidate mutations, but they struggle to compare variants between different genes on the same scale.
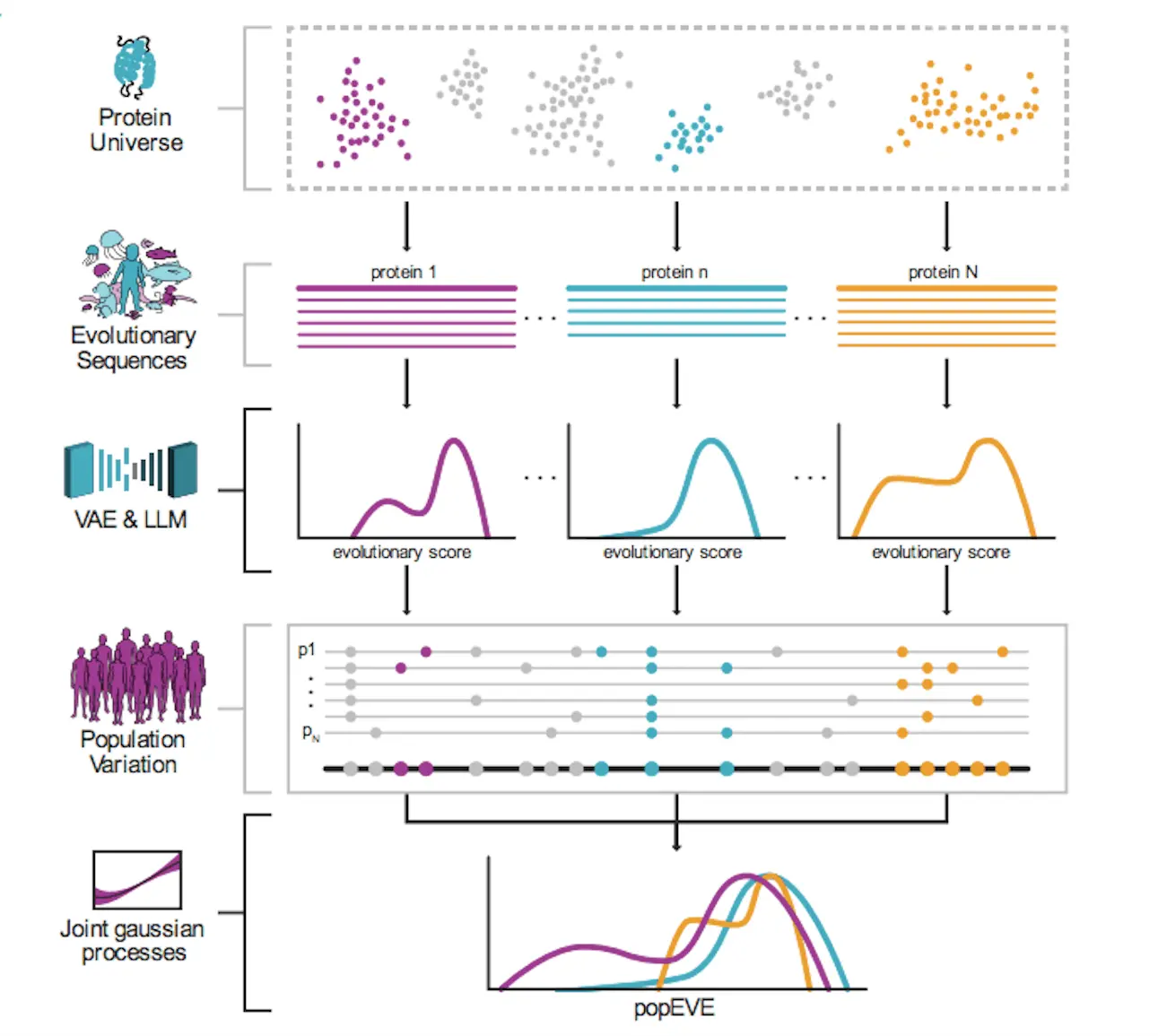
To address this limitation, the researchers developed popEVE – a revolutionary new AI technique that ranks every possible amino acid mutation across the entire human proteome on a unified scale of pathogenicity. This enables comparing the likely harmfulness of mutations between genes to pinpoint the variants most likely involved in a patient’s disease.
Combining Evolutionary Data and Population Genetics
PopEVE leverages both evolutionary conservation data and population-level genetics to achieve accurate variant effect predictions that generalize across the whole proteome.
The model incorporates mutation sensitivity profiles from a technique called EVE (evolutionary model of variant effect), which analyzes patterns of amino acid substitutions over evolutionary timescales to reveal positions in proteins most important for function. However, evolutionary constraints alone don’t necessarily predict consequences at an organism level.
To transform EVE’s functional disruption scores into a pathogenicity measure tied to human health impacts, popEVE also integrates data on genetic variation in the UK Biobank. The AI model learns a mapping that calibrates EVE scores based on whether variants are observed across Biobank participants’ genomes.
This grounds predictions in terms of human-specific fitness effects – pushing scores towards neutrality unless there is compelling evidence a mutation is detrimental.
State-of-the-Art Performance in Diagnosing Rare Diseases
The researchers tested popEVE’s ability to pinpoint disease-causing mutations by applying it to de novo (spontaneously arising) variants from over 31,000 patients with undiagnosed developmental disorders.
Remarkably, popEVE achieves a 15x enrichment for pathogenic variants in this cohort over background mutation rates – significantly outperforming existing methods. The model also shows the best performance at separating cases from healthy controls.
These results demonstrate popEVE’s power to rank mutations by their likelihood of disrupting health, even for never before seen variants in genes with no previous disease association.
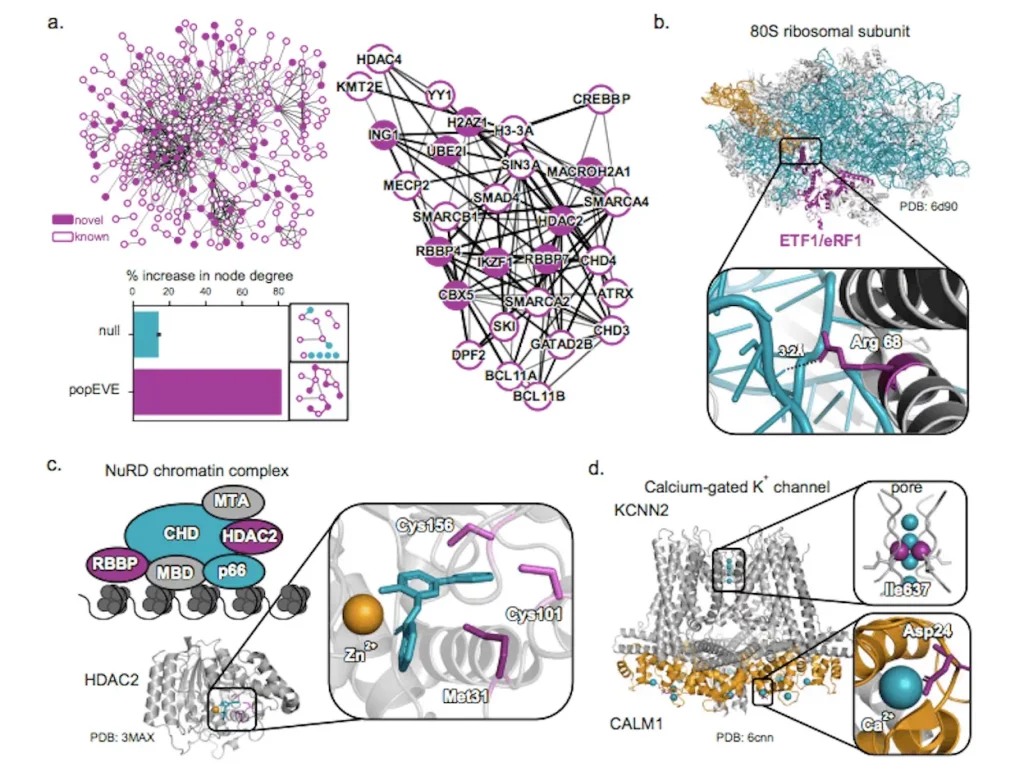
Image Source: https://doi.org/10.1101/2023.11.27.23299062
Accelerating Discovery of Novel Disorders
In total, popEVE implicates variants in 442 genes as probable drivers of the developmental disorders in the study cohort. Intriguingly, over half of these gene associations are completely novel, undetected by previous analyses.
By comparing mutation severity across the proteome, popEVE can pinpoint likely causal candidates even for extremely rare “single-patient” disorders too unusual to find by recurrence. The researchers provide extensive functional evidence that these novel genes interact with known players in developmental biology and contain mutations disrupting critical protein regions.
Overall, the model nominates 119 previously unknown gene-disorder relationships. The researchers emphasize these may indicate innovative treatment possibilities if the precise genetic basis can be established.
Harnessing Evolution to Understand Human Disease
PopEVE underscores the immense value of evolutionary conservation patterns for revealing the links between genotype and phenotype. Nature’s grand protein engineering experiment provides an unmatched vantage into subtle genetic underpinnings far beyond what can be inferred from human variation alone.
This research illustrates the power of merging these evolutionary insights with large-scale human sequencing efforts. It serves as a blueprint for how similar AI techniques may accelerate the understanding of diverse medical mysteries.
The scientists suggest popEVE shifts the paradigm for genetic testing interpretation – away from binary classification of mutations as pathogenic or benign towards a more nuanced spectrum reflecting real-world variation in disease outcomes.
Conclusion
By contextualizing mutations relative to the entire proteome, the model opens new clinical possibilities to assess genetic contributions even for conditions unheard of in medical literature. As the authors say, “Patients with unique sets of symptoms and genotypes would still go undiagnosed,” unless we have a deeper view of genetic variation across billions of years of evolution.
Story Source: Reference Paper | popEVE is available at https://pop.evemodel.org/
Important Note: medRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:



Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











{kind=link}