
RNA sequencing is currently a useful tool for studying the dynamic transcriptional mechanisms that control the operation of eukaryotic cells. Nevertheless, complicated experimental designs with various biological variables and multiple analysis time points cannot be automatically analyzed by the tools that are currently on the market for the analysis of raw sequencing data. Multiple programs are combined into a single framework by the MultiRNAflow suite by researchers from the University of Lorraine, France, enabling supervised and exploratory statistical analysis of temporal data for various biological situations.
Introduction
Nuclear DNA genes in eukaryotic cells are translated into messenger RNA molecules prior to being translated into proteins that maintain normal cellular activities. Stochastic events that impact transcription during cell dormancy cause a transcriptional noise within the cell.
Thousands of genes are triggered upon changes to the cellular environment (such as receptor activation or cellular stress), which causes a dynamic, transient transcriptional response that enables the cells to adjust to the initial environmental disturbance. Biologists investigate changes in these temporal transcriptional responses, which can result in diseases like cancer, in great detail using occasionally intricate experimental setups. Thanks to recent advances in technology, it is now possible to use RNA sequencing (RNAseq) to measure the transcription of every gene in the genome.
Understanding MultiRNAflow
Biologists and bioinformaticians who want to automatically analyze RNAseq datasets multiple times and under multiple biological conditions should use the MultiRNAflow software. Following an unsupervised data analysis phase, the typical questions that our package can address are: selecting differentially expressed (DE) genes specific to a given biological condition or time, for example, in order to infer a gene network model specific to that biological condition; or performing functional and gene ontology (GO) analyses of genes specific to a biological condition, for example, in order to identify genes specific to a biological condition that are involved in a given cellular program (e.g., proliferation of cancer cells).
Tools like IDEAL, RNASeqR, SeqGSEA, and RNAflow are among the R packages that offer methods for data normalization, enabling unsupervised analysis, and locating DE genes. These tools make use of DESeq2 and EdgedR to do the normalization and DE analysis. While RNASeqR can only identify DE genes in samples from two biological circumstances, the others can all identify DE genes in samples from various biological conditions. They also conduct GO enrichment analyses of them. Notwithstanding their potential for adaptation, these packages were not intended to handle temporal data. However, none provides an automated and unified framework for RNA-seq data analysis with many biological circumstances and multiple time points.
The MultiRNAflow suite unifies methodological tools from different packages into a single framework, enabling the following tasks:
- Exploratory (unsupervised) data analysis.
- Supervised statistical analysis of dynamic transcriptional expression (DE genes), utilizing the DESeq2 package.
- Functional and GO analyses of genes using gProfiler2 and file generation for additional analyses with multiple software programs.
Methodology
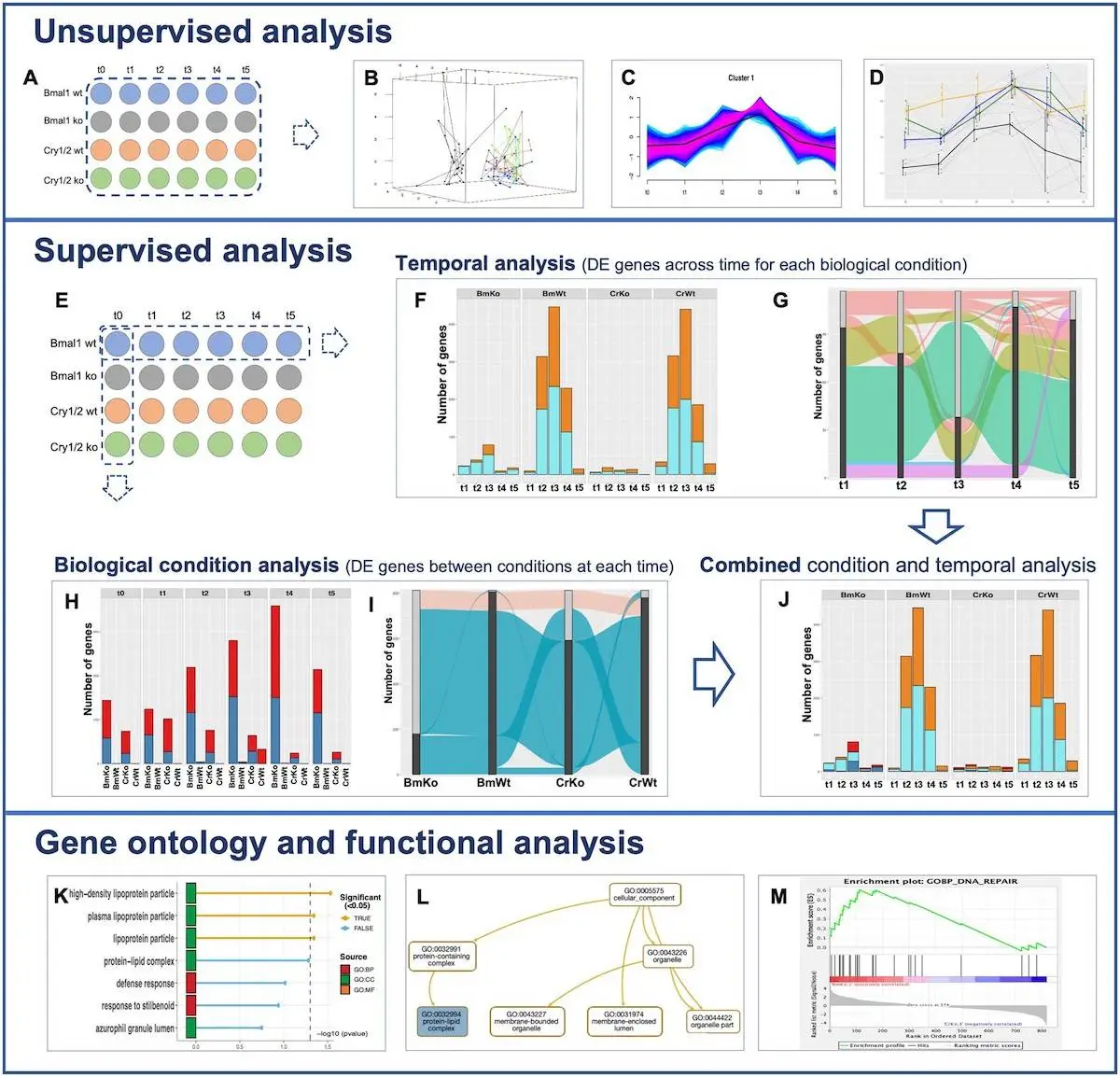
Transcriptional RNAseq raw count data from an experimental design with multiple conditions and numerous times is supported by MultiRNAflow. The reference time, denoted as t0 in the experimental design, is assumed to be different from other times, recorded as t1 to tn, corresponding to a collection of reference measurements. Several graphical outputs are provided by the software, including a dataset of the genes Cry1/2 and Bmal1 on mouse transcriptional dynamics. Each of the four biological situations and six-time points on the experimental map has four duplicates. Three additional datasets with various experimental designs are provided in the package description, and further outputs are supplied in the extra material.
Data Analysis in MultiRNAflow
Principal component analysis, or PCA, is used to do a factorial analysis of the temporal transcription of replicates for all biological circumstances. A dynamic 3D PCA is used to optimize the visualization. It is also necessary to carry out a hierarchical clustering on principal components (HCPC). One way to aggregate samples based on the expression levels of individual genes is through a hierarchical clustering of samples versus genes using scaled expression data. It is possible to group replicates and biological situations with comparable transcriptional behavior using hierarchical clustering of data based on correlations. Within a sample, groups of genes exhibiting more frequent temporal behavior are highlighted using unsupervised clustering of temporal gene expression using Mfuzz for each condition. Additionally, a gene of interest’s temporal expression profile inside a particular cluster can be shown thanks to the package’s graphical features.
Conclusion
Researchers have created the MultiRNAflow software to analyze RNAseq datasets with various biological circumstances and multiple time points. It facilitates inferring gene network models, conducting functional and gene ontology (GO) investigations, and undertaking unsupervised analysis of differentially expressed (DE) genes unique to a particular biological situation or period. The MultiRNAflow suite offers a cohesive framework for unsupervised analysis, supervised statistical analysis of DE genes, and functional and GO analyses of genes, in contrast to existing R packages that employ DESeq2 and EdgedR for normalization and DE analysis. Additionally, it creates files that can be analyzed further using different programs.
Article Source: Reference Paper | MultiRNAflow’s R package is freely available on Bioconductor | The latest version of the source code is available on GitHub.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}